Hány SARS-CoV-2-vírustörzsről tudunk?

„Csak a változás örök” – mondta Hérakleitosz, és abban egészen biztosak lehetünk, hogy nem a virális genomok evolúciójára gondolt. Ettől még azonban a mindennapjainkat felforgató koronavírus, a SARS-CoV-2 genomjának kontextusában is igaz ez a mára már kicsit közhelyes gondolat.
Bár a vírusok a legtöbb erre szakosodott kutató szerint nem tekinthetők a szó szigorú értelmében „élő szervezeteknek”, örökítőanyaguk változására mégis ugyanazok a szabályszerűségek vonatkoznak. És az élő szervezetek egyik jellegzetessége, hogy az örökítőanyagukat tökéletlenül másolják – pont ez adja azt a genetikai variabilitást, ami a természetes szelekció és ezáltal az egész evolúció alapanyagául szolgál, amely kis számú még fittebb (és nagy számú kevésbé fitt) egyed létrehozásával egyengeti az utat a környezethez való optimálisabb alkalmazkodás felé.
Nincs ez másképp a SARS-CoV-2 durván 30 ezer bázispárra rúgó genomja esetében sem. Minden egyes másolás (és csak egyetlen megfertőzött sejt esetében rengeteg ilyen lehet) magában hordja annak az esélyét, hogy az RNS-alapú örökítőanyag másolását végző polimeráz enzim téved, vagy valamilyen sejten belüli mechanizmus mutációt hoz létre, és az új virális genom egy vagy ritkábban több ponton különbözni fog a „szülői” RNS-től. Ha egy ilyen mutáció a vírus számára fontos fehérjében okoz valami káros változást, márpedig ez a gyakoribb, akkor a következő sejt megfertőzésekor a vírus nem tud hatékonyan replikálódni, így ez a vonal előbb-utóbb elhal. Ha a mutációnak nincs hatása (szakzsargonban: neutrális), akkor fennmaradhat, de ez a szerencsén múlik, hiszen a neutrális mutációt hordozó vírusnak még időben kell egy új gazdaszervezetet találnia, ahol szaporodni kezdhet. És végül, a legritkább esetekben az is előfordulhat, hogy a egy olyan mutáció jelenik meg, amitől a vírus jobban fog tudni szaporodni: gyorsabban replikálódik, vagy hatékonyabban fertőz meg új sejteket/gazdaszervezeteket. Ha ilyen lényegi változás jelenik meg, akkor egy új vírustörzsről beszélhetünk, addig az egyes mutációk alapján „kládokat” különböztethetünk meg.
Az eddig világszerte megszekvenált sok ezer SARS-CoV-2-genom egymáshoz vetése alapján két dolog fog feltűnni, szinte azonnal. Egyrészt az, hogy a vírus genomja nem meglepő módon folyamatosan változik. Ez a változás nem túl gyors: a mostani sebesség mellett kb. 24 mutáció jelenik meg a kiindulási genomhoz képest egy év alatt. Vagyis durván kéthetente rögzül egy-egy új mutáció, ami a fent leírtakat figyelembe véve kifejezetten komótos változásnak tűnik; lényegesen lassabb, mint amit az influenza, vagy a HIV esetében tapasztalunk.
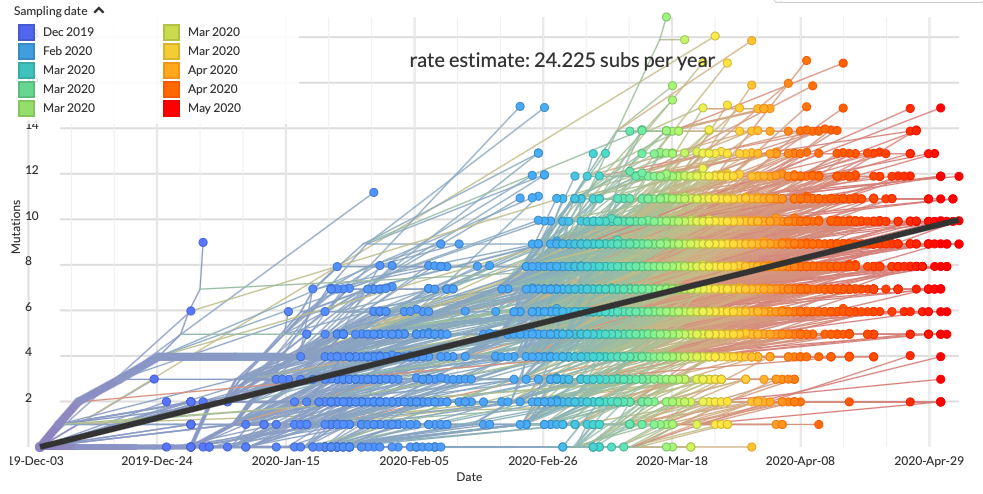
A másik érdekesség, hogy a genomok eléggé egyformák, igazából – egyelőre – csak kevés helyen különböznek, ami azt valószínűsíti, hogy ez a vírus csak egyszer, valamikor 2019 november végén, vagy december elején ugrott át emberre.
Persze a hasonlóság még nem jelenti azt, hogy csak egy törzs van, és ezzel elértünk napjaink egyik népszerűnek tűnő szabadidős elfoglaltságához: annak megvitatásához, hogy léteznek-e „durvább” SARS-CoV-2 koronavírus-törzsek, és ha igen, ennek tulajdonítható-e, hogy egyes országokban, például Olaszországban a halálozási adatok sokkal drámaibbak lettek, mint például Kínában. Az egészen rövid válasz az, hogy a legtöbb virológus nem lát erre bizonyítékot.
A vírus természetesen folyamatosan változik, ezért időről időre indokolt is megvizsgálni, hogy esetleg már kialakult-e valahol egy új törzs. Az eddig rendelkezésre álló adatok szerint ugyanakkor még csak kládokról beszélhetünk. Igazából, ahogy a posztban használt grafikonok forrása, a Nextstrain adatbázis egyik atyja, Trevor Bedford részletesen kifejtette, eddig egyetlen új mutációnál merült fel komolyabban, hogy annak következtében alakult ki a vírus picit gyorsabb szaporodását okozó változás, de egyelőre ezt nem sikerült igazolni. Sőt a legújabb eredmények fényében az a valószínűbb, hogy inkább csak a sejtek belső védekező mechanizmusa által újból és újból létrehozott változásról van szó.
Mi a helyzet a Magyarországon terjedő vírussal?
De mit tudunk a Magyarországon keringő vírusról mondani? Az erre vonatkozó információink a Pécsi Tudományegyetemen működő kutatócsoport heroikus munkájának köszönhetők. Az eddig regisztrált több mint 3600 hazai betegből 32 esetben történt meg a vírus izolálása és szekvenálása. Ez kicsit kevesebb, mint egy százalék, ami kevésnek tűnhet, de igazából nem az, sőt a térségben kifejezetten jó értéknek számít. A visegrádi országok közül a csehek és lengyelek hasonló nagyságrendű, de kevesebb genomot szekvenáltak meg, míg déli és keleti szomszédaink nagyságrendileg kevesebbet, a lényegesen jobb erőforrásokkal rendelkező Ausztria pedig 142-t. Fontos azt is hozzátenni, hogy bár természetesen a több adat itt is biztosan informatívabb, egy idő után az új szekvenciákból arányosan kevesebb új információ nyerhető, így indokolt lehet az emberi és anyagi erőforrások más irányba terelése. (Információink szerint ugyanakkor ilyenre itthon nem volt szükség, így várható a magyar adatok további bővülése.)
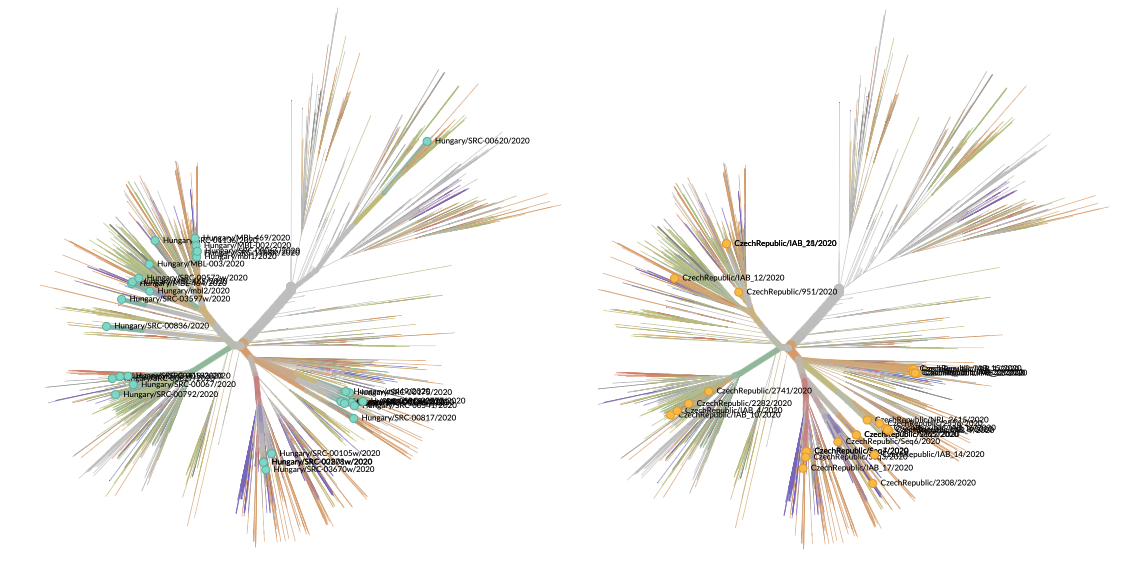
A kérdés inkább úgy tehető fel, hogy mennyire lehet reprezentatív a 32 minta az országban keringő összes vírusra nézve? Kicsit úgy lehet erről is gondolkozni, mint arról, hogy mennyi annak a valószínűsége, hogy egy folyóparton fekvő 3600 kavicsból 32 felszedése jó képet ad a teljes fövenyről. A véletlenszerű mintavételezésnek könyvtárnyi irodalma van, amit sokszor alkalmaznak is – hogy ne menjünk messzire, a H-UNCOVER felmérés is ezt használta a vizsgálandó alanyok kiválasztásához –, a 32 izolátum esetében erre nem volt igazán eddig lehetőség, így egy kicsit bizonytalanabb terepen mozgunk. A kirajzolódó kép azonban összességében egybevág azzal, amit a térség más országaiban, például Csehországban is látunk: a vírus elsősorban Nyugat-Európából, feltehetőleg a határok lezárását megelőző hazautazási hullámmal jutott Magyarországra. A jelek egyértelműen azt támasztják alá, hogy több esetben spanyol, francia és brit területeken terjedő vírusokkal rokon patogének jelentek meg itthon is, ami független behozatali eseményekre utal. (A járvány első hazai hullámában megbetegedő iráni diákok mintái nincsenek a megszekvenáltak közt, és az, hogy ezekkel rokon mintákat sem látunk, optimizmusra ad okot arra nlzve, hogy annak a behozatali eseménynek tényleg sikerült véget vetni.)
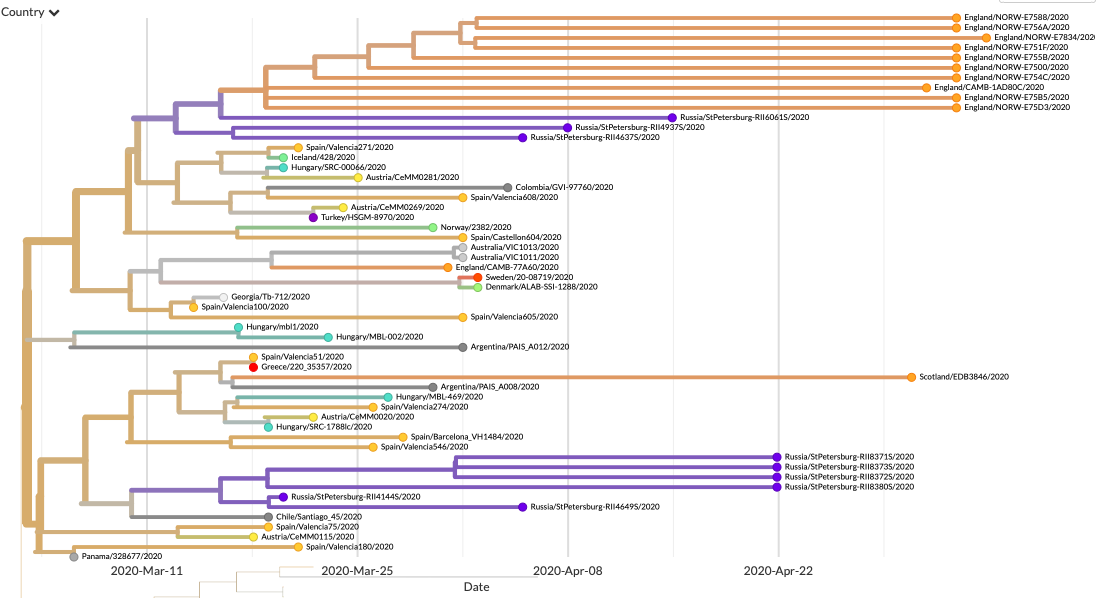
Szintén megfigyelhető, hogy már ebben a 32 mintában is látjuk nyomait a hazai terjedésnek, vagyis a minták nagyon közeli rokonságban vannak egymással (pár esetben meg is egyeznek a szekvenciák). Ez természetesen nem meglepő: egyéb adatok alapján tudjuk, hogy kórházakban és az idősotthonokban így terjed a vírus, és a pécsi kutatók mintáinak egy része is „kórházi klaszterekből” származik. Viszont azt is sejteti, visszatérve az előző gondolatra, hogy a mintavétel aligha tekinthető véletlenszerűnek.
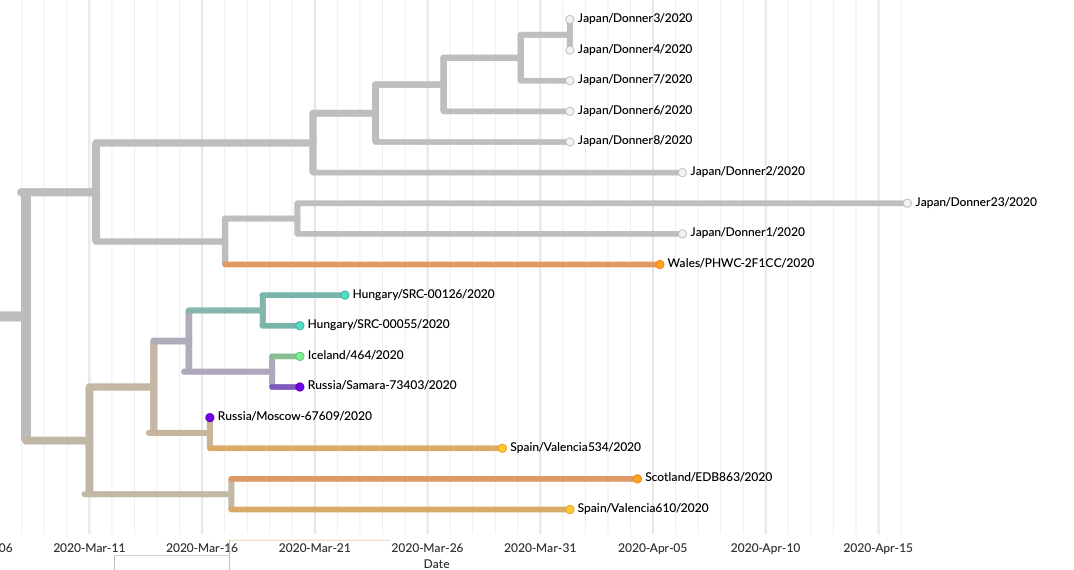
Márpedig ha van valami, amin lehetne változtatni a jövőben, az talán pont ez. Most, hogy a járvány első, erősen elfojtott hullámán túl vagyunk, érdemes végiggondolni, mi az, amin változtatnunk kell a még hatékonyabb védekezéshez. Ennek a hullámnak a hazai enyhe lefolyása időt és lehetőséget adott arra, hogy felkészüljünk a következő hónapokra, és ennek része kell legyen a vírus terjedésének monitorozása.
A klasszikus PCR-alapú tesztek információt adnak a vírus jelenlétéről egy vizsgált beteg vagy tüneteket nem mutató személy szervezetében, illetve a szennyvíz vizsgálata esetén arról, hogy egy adott közösségben jelen van-e a vírus. De ezek a vizsgálatok a természetüknél fogva egyáltalán nem, vagy csak részlegesen tudnak információt adni a vírus változásáról. Ehhez továbbra is elengedhetetlenek a megfelelően megtervezett mintavételezésű szekvenálás-alapú elemzések, amelyek a későbbiekben azt is segíthetnek majd kimutatni, ha a terjedő víruskládok közül valamelyik valóban egy új törzzsé alakul.
(A poszt ábrái a nextstrain.org-ról származnak. Köszönet Ari Eszternek a szöveg lektorálásáért.)
Kapcsolódó cikkek a Qubiten: