A mesterséges intelligencia végre feltárhatja a fehérjék szerkezetének rejtélyeit

A fehérjék az élethez szükséges kulcsfontosságú makromolekulák, amelyek kémiai reakciókat katalizálnak, lehetővé teszik az örökítő anyagban tárolt genetikai információ kifejeződését és másolását, valamint szállító, strukturális, jelátviteli, mozgató és egyéb funkciókat látnak el.
Építőelemeik az aminosavak, amelyek a genetikai kód által meghatározott sorrendű láncban alkotják a fehérjéket. A legtöbb fehérje a genetikai kód által meghatározott 20 aminosavból épül fel, amelyek kovalens típusú peptidkötéssel kapcsolódnak egymáshoz, és egy polipeptid láncnak nevezett egyenes struktúrát képeznek. Ez egy funkcionális, háromdimenziós szerkezetet hoz létre a fehérje feltekeredésnek nevezett folyamat során.
A fehérjék felépítéséhez szükséges információ végső soron a DNS-ben tárolódik. Innen az információ előbb a transzkripció folyamatában mRNS-é íródik át, majd az mRNS a transzláció folyamán, a riboszóma segítségével lefordításra kerül aminosavsorrendé, létrehozva a polipeptid láncot. Ez még további, úgynevezett poszttranszlációs módosításokon mehet keresztül, de végső, biológiai funkcióját csak a fehérje-feltekeredési folyamat után nyeri el, ami általában már a transzláció során megkezdődik. Bár a fehérjék a genetikai információ által meghatározott alakú, háromdimenziós struktúrának köszönhetik funkciójukat, a biokémia talán egyik legnagyobb megoldatlan problémáját jelenti ennek előrejelzése a fehérjék szekvenciájából, azaz aminosavsorrendjéből.

A fehérjék térszerkezetének előrejelzése nemcsak elméleti szempontból fontos, hanem forradalmasíthatja több, valószínűleg fehérjék hibás feltekeredése által okozott neurodegeneratív betegség kezelését és a biotechnológiai iparágat is. Szerkezetük pontos meghatározása jelenleg komplikált, időigényes és drága módszereket (pl. röntgen-krisztallográfia) igényel. Ezen azonban hamarosan változtathat a londoni székhelyű, most a Google anyacége, az amerikai Alphabet tulajdonában álló vállalat, a DeepMind AlphaFold szoftvere és más, gépi tanulási és mélytanulási módszereket felhasználó megoldások.
Az AlphaFold mindent letarol
Ahhoz, hogy megértsük a mesterséges intelligenciát alkalmazó modellek és algoritmusok jelentőségét a fehérjeszerkezet előrejelzésben, érdemes pár, a területen gyakori fogalmat definiálni:
A gépi tanulást (machine learning) alkalmazó algoritmusok és modellek explicit utasítások nélkül képesek adatmintázatokból következtetéseket levonni. Ehhez „tréningadatok” alapján építenek egy matematikai modellt, amelynek segítségével előrejelzéseket tudnak adni más hasonló adatokra is.
A mélytanulás (deep learning) egy gépi tanulási módszer, amely biológiai rendszerek által inspirált, mesterséges neurális hálózatokon alapszik. Ezek több rétegből állnak, és egyre magasabb szintű, komplikáltabb problémák megoldására képesek. Egyik altípusuk a mély neurális hálózatok (deep neural networks).
Ezeknek a módszereknek köszönhetően 2018 az áttörés éve volt a fehérjeszerkezet előrejelzésben:
Tavaly decemberben rendezték meg ugyanis a 13. CASP fehérjeszerkezet-predikciós versenyt, amelyet a DeepMind AlphaFold (A7D) programja nyert meg az új fehérjefeltekeredéseket előrejelző kategóriában, messze lekörözve vetélytársait. A DeepMind mesterségesintelligencia-programok fejlesztésével foglalkozik. Legnagyobb dobása eddig az AlphaGo, egy gépi tanuláson alapuló szoftver volt, amely 2016-ban öt alkalomból négyszer legyőzte góban az egyik legjobb játékost, a dél-koreai Lee Sedolt. A területtel foglalkozó szakemberek szerint ez egyértelmű mérföldkő volt a mesterségesintelligencia-kutatásban, néhányan pedig az általános mesterséges intelligencia felé tett lépésnek tekintik az AlphaGót.
A DeepMind az AlphaGo mellett 2016-ban kezdett foglalkozni a fehérjefeltekeredés problémájával. Az AlphaFoldnak elnevezett szoftver két különböző fehérjetulajdonságot állapít meg azok genetikai kódjából, mély neurális hálózatokon alapuló technológiával. Az első ezek közül az aminosavpárok közötti távolságok, a második pedig az ezeket az aminosavakat összekapcsoló kémiai kötések közötti szögek meghatározása.
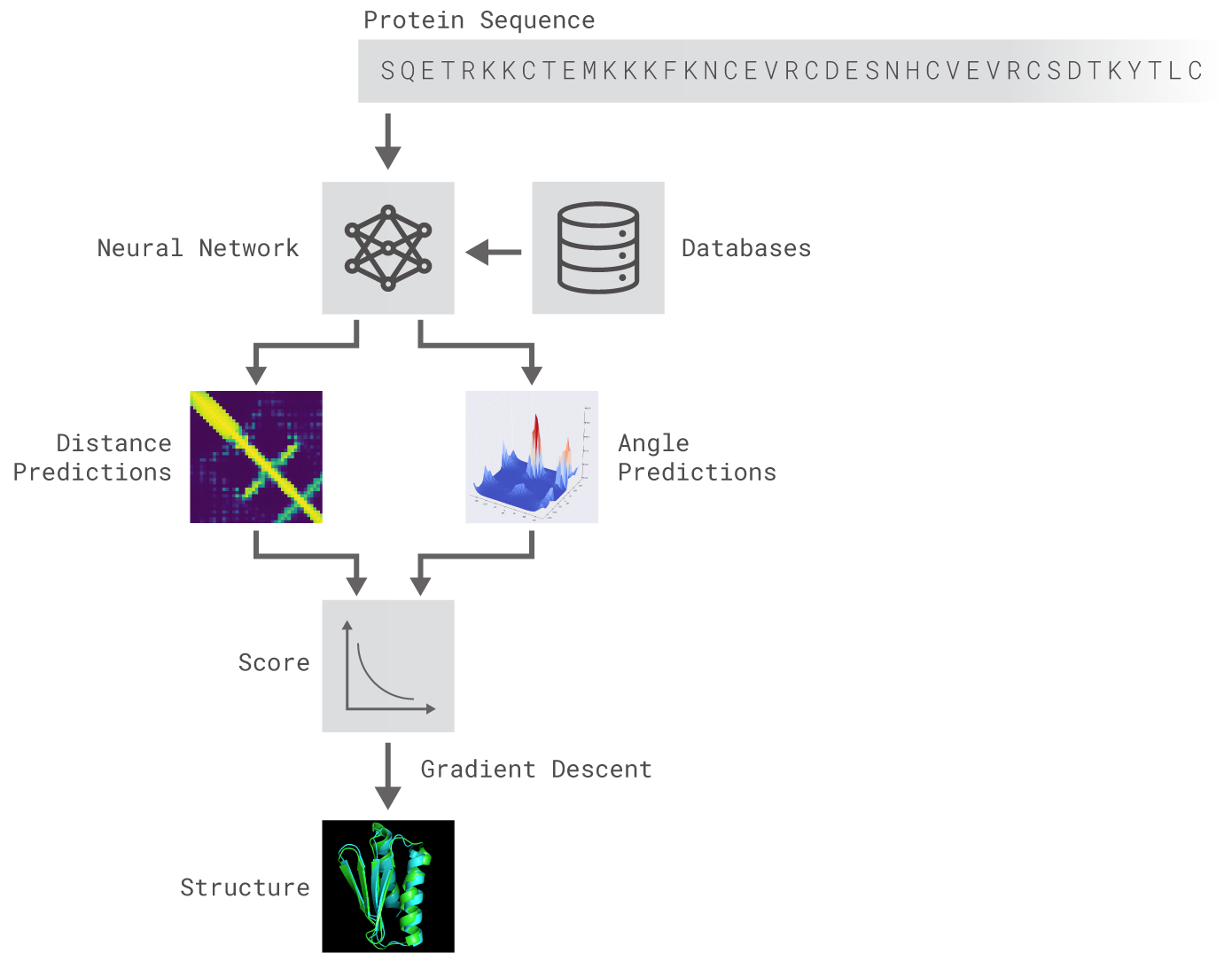
Az első neurális hálózatot egy fehérjeszerkezeti adatbázisból kiválasztott fehérjéken gyakorlatoztatták, majd a modell előrejelzését a meglévő referenciával összevetve egy valószínűségi pontszámot generáltak a lehetséges struktúrákhoz. A második neurális hálózat direktben egy pontszámot adott ki a fehérje geometriai jellemzői és az első hálózat távolságpredikciói alapján. A gradient descent nevű, gépi tanulásban általánosan használt matematikai eljárást felhasználva a szakemberek optimalizálták a pontszámokat, ami pontosabb térszerkezeteket eredményezett.
A terület egyik szakértője, Mohammed AlQuraishi rendszerbiológus a Bioinformatics folyóiratban értékelte az AlphaFold CASP13-on aratott sikerét, amelyet szerinte a megfelelően megválasztott biokémiai koncepciók és korszerű gépi tanulási módszerek tettek lehetővé. AlQuraishi komoly előrelépésnek nevezte a modellt, és felvetette annak lehetőségét, hogy a trendek alapján 5 éven belül a a jelenlegi 55 százalékos pontosság helyett 85 százalékos is elérhető lesz az alkalmazásával. Hozzátette, hogy még az AlphaFold nélkül is egyértelmű a terület korábbiaknál gyorsabb ütemű fejlődése, amely részben a mélytanulási módszerek elterjedésének köszönhető.
Egymilliószor gyorsabb szerkezet-meghatározó program
Áprilisban AlQuraishi egy új, a korábbiaknál sokkal gyorsabb fehérje szerkezetet előrejelző programot tett közzé. A Cell Systems rendszerbiológiai folyóiratban publikált tanulmányában részletesen bemutatta mélytanulást alkalmazó modelljét, amely kombinálja a fehérjeszerkezet lokális és globális jellemzőit. Utóbbi a teljes térszerkezet megállapításának irányába optimalizál, a kémiai kötések jellemzőinek tiszteletben tartásával. A modell lényegében folyamatos ismétlésen keresztül, önmagát javítja annak érdekében, hogy megtanulja, milyen matematikai kapcsolatban áll a fehérje szekvenciája a struktúrájával.
A mélytanulási modell meghatározza az aminosavakat összekapcsoló kémiai kötések legvalószínűbb szögét, valamint a kötések körüli elfordulási szöget, amely kontrollálja, hogyan viszonyul egy lokális struktúra az egész szerkezethez. Ezt sokszor, több ezer különböző fehérjén megismételve és az eredményeket az ismert struktúrákkal összehasonlítva a modell tréningezhető.
A modellt két különböző módon is tesztelték: első körben új tekeredési mintázatokat kellett előrejeleznie, és ebben páratlanul pontosnak bizonyult. A másodikban már ismert tekeredési mintázatokat kellett kiadnia, szerkezeti sablonok nélkül, és ebben majdnem olyan jól teljesített, mint a legjobb kísérleti adatokon alapuló fehérjeszerkezeti sablonokat használó modellek.
AlQuraishi szerint a bizonyos helyzetekben a korábbiaknál egymilliószor gyorsabb modell még tovább fejleszthető, és felhasználható lehet új gyógyszerek felfedezésében és fehérjék tervezésénél is. Ehhez azonban a jelenlegi 60 nanométeres helyett 10-20 nanométer pontosságot kell elérnie, amivel ténylegesen feltárhatja a fehérjék teljes atomi szerkezetét.
Fehérjetervezés neurális hálózatokkal
A Heidelbergi Egyetem és a berlini egészségügyi intézet közös kutatómunkája eredményeként létrehozott DeeProtein algoritmus a fehérjeszekvenciából képes meghatározni a fehérjék pontos sejtbeli funkcióját. A Nature Machine Intelligence folyóiratban idén májusban közzétett tanulmányukban a kutatók ezt a neurális hálózatot és lehetséges felhasználásait mutatják be. Más ilyen típusú szoftverekhez hasonlóan a program működése – pontosabban az, hogy miért rendel bizonyos szekvenciákhoz egy adott funkciót – ismeretlen marad. A szakemberek célja tehát az volt, hogy feltárják a program előrejelzései mögött meghúzódó okokat.
Ehhez innovatív módon egy gépi tanulással foglalkozó szakemberek által kifejlesztett, úgynevezett „kitakarási érzékenységvizsgálat” eljárást használtak. Ennek segítségével sikerült a fehérjék biológiai funkciója szempontjából lényeges pozíciókat meghatározniuk a fehérje szekvenciákban. Így pedig fontos kötő alegységeket és kémiai reakciók szempontjából aktív területeket is ki tudtak mutatni a vizsgált fehérjékben.
A DeeProteint több fehérje példán tesztelve sikeresen bizonyították, hogy az alkalmazott érzékenységi analízis technika alkalmas lehet a fehérjék funkcióinak vizsgálatára, és később akár fehérjemérnökségre is.
Messze még a cél
Fontos leszögezni, hogy messze járunk még attól, hogy az eddigi eredményeket a gyakorlatban is fel lehessen használni. Ahhoz, hogy megértsük ennek okát, vissza kell kanyarodnunk a CASP13 konferenciához és az AlphaFoldhoz. AlQuraishi a konferenciát értékelő vélemény cikkében és blogbejegyzésében is kifejti, hogy bár az AlphaFold nagyon jól teljesít a nagyvonalú szerkezet meghatározásában, a praktikus alkalmazásokhoz szükséges nagyobb felbontású előrejelzései már sokkal rosszabb, mindössze 40 százalék körüli pontosságúak.
Kérdés még az is, hogy alapvetően akadémiai vagy mérnöki-számítástechnikai terület lesz-e a fehérjestruktúra-előrejelzés a jövőben, illetve az is, hogy miként alkalmazkodnak majd a DeepMind sikeréhez az akadémiai kutatók.
Az viszont teljesen világos, hogy a fehérjefeltekeredés problémájának megoldásához a mesterséges intelligencia és a nagy mennyiségben rendelkezésre álló biológiai adat kombinálásán keresztül vezet az út.