Áttörést hoz a biológiába a minden eddiginél pontosabb fehérjekutató algoritmus, az AlphaFold2

Évtizedek óta a molekuláris biológia egyik legfontosabb célja a földi élethez nélkülözhetetlen fehérjék térbeli szerkezetének feltárása, és annak megismerése, hogy miként jön létre egy fehérje a feltekeredésnek nevezett folyamat során, egymás utáni aminosavak láncolatából háromdimenziós struktúrává. A biológiai folyamatok megértéséhez, gyógyszerek felfedezéséhez és biotechnológiai áttörésekhez elengedhetetlen a fehérjék szerkezetének ismerete, kísérleti módszerekkel azonban eddig több milliárdból csupán hozzávetőleg 100 ezer fehérje térszerkezetét sikerült nagy erőfeszítések árán meghatározni.
2020-ban a mélytanulást (deep learning) alkalmazó mesterséges intelligencia forradalmi áttörést hozott ezen a területen is. A Google anyacége, az Alphabet DeepMind nevű leányvállalatának AlphaFold2 algoritmusa soha nem látott, kísérleti módszerekkel vetekedő pontosságot ért el a fehérjék szerkezetét számítási módszerekkel előre jelző CASP14 versenyen, ahol minden más versenyzőt maga mögé utasított.
A fehérjékről minden meglévő strukturális, genetikai és evolúciós információt felhasználó AlphaFold2-t 2021 júliusában írták le a DeepMind kutatói a Nature folyóiratban, a GitHub-on pedig mindenkinek nyíltan elérhetővé tették. Ugyanekkor, szintén a Nature-ben ismertették az emberi sejtek által kifejezett összes fehérjéhez, az emberi proteomhoz tartozó térszerkezetek feltárását, amelyeket főbb genetikai modellélőlények proteomjával együtt, az Európai Bioinformatikai Intézettel (EBI) együttműködésben publikáltak.
Ezzel egy csapásra több százezer fehérjéhez tartozó, az AlphaFold2 által meghatározott térszerkezet vált elérhetővé a biológusoknak, a részben az EBI által létrehozott nemzetközi fehérje-adatbázisban, a UniProtban. Az azóta eltelt több mint 6 hónap elegendő volt ahhoz, hogy a tudományos közösség a gyakorlatban is kipróbálhassa, mennyiben segíti a molekuláris biológiai és biotechnológiai kutatásokat az új módszer.
Már a gyógyszerkutatásban is részt vesz az AlphaFold2
Az új algoritmushoz fűzött reményeket jól érzékelteti Patrick Cramer német biokémikusnak a Nature Structural & Molecular Biology folyóiratban tavaly augusztusban publikált véleménycikke, amelyben áttörő eredménynek nevezte az AlphaFold2-t. Az algoritmus szerinte praktikusan megoldja a fehérje feltekeredési problémát, még akkor is, ha elméleti szinten továbbra sem igazán értjük, hogyan kódolja az aminosavsorrend a fehérjék háromdimenziós szerkezetét. Cramer ugyanakkor arra is felhívta a figyelmet, hogy a módszer nem kiváltja a jelenleg használt kísérleti térszerkezet-meghatározó módszereket, hanem kiegészíti és segíti majd azokat.
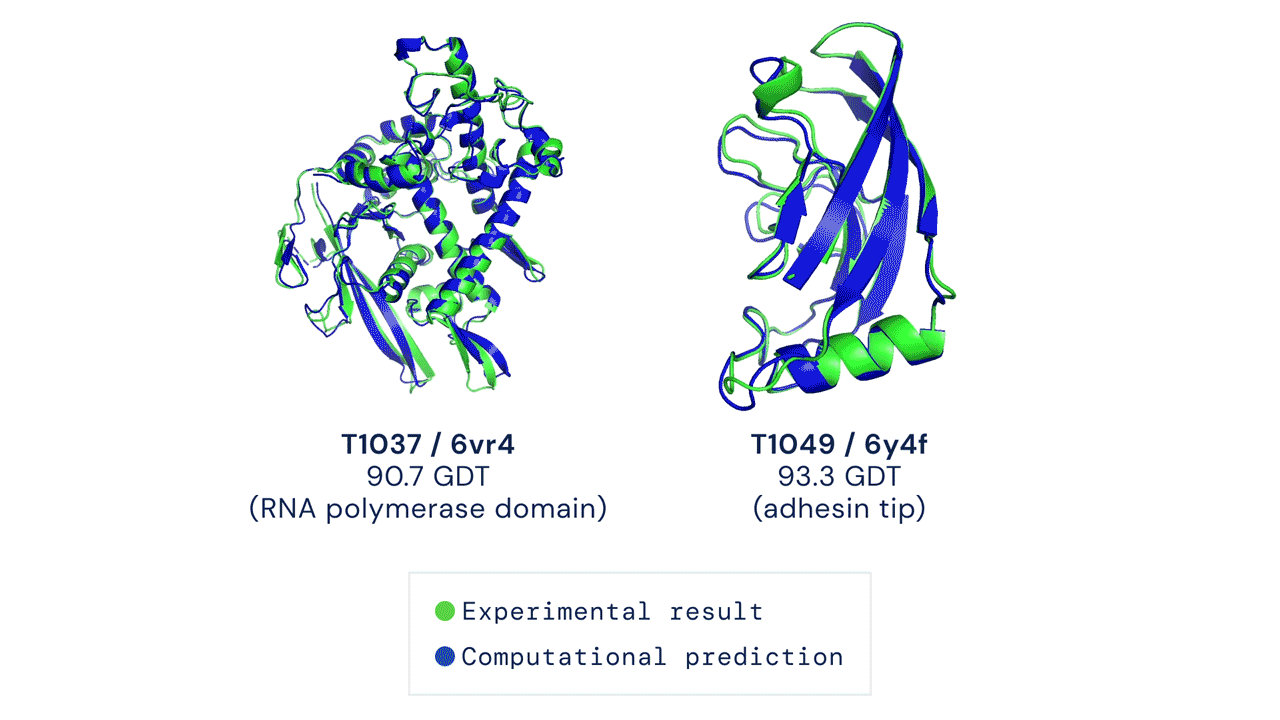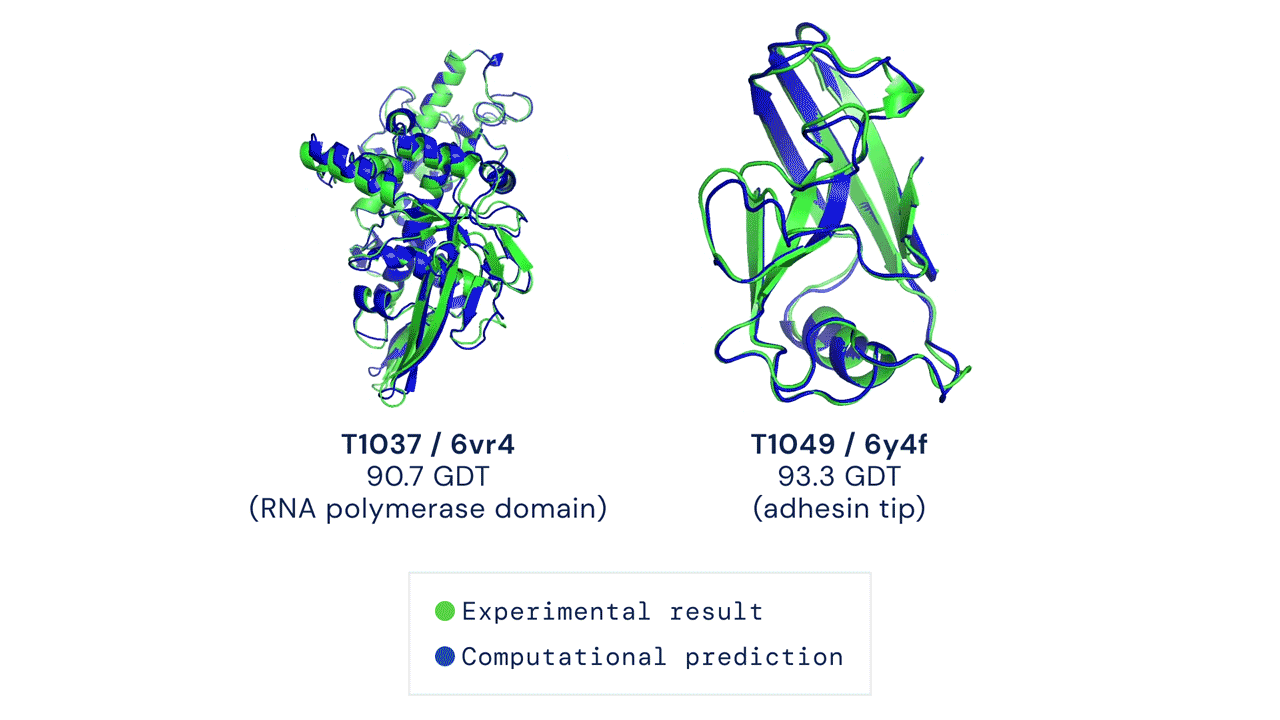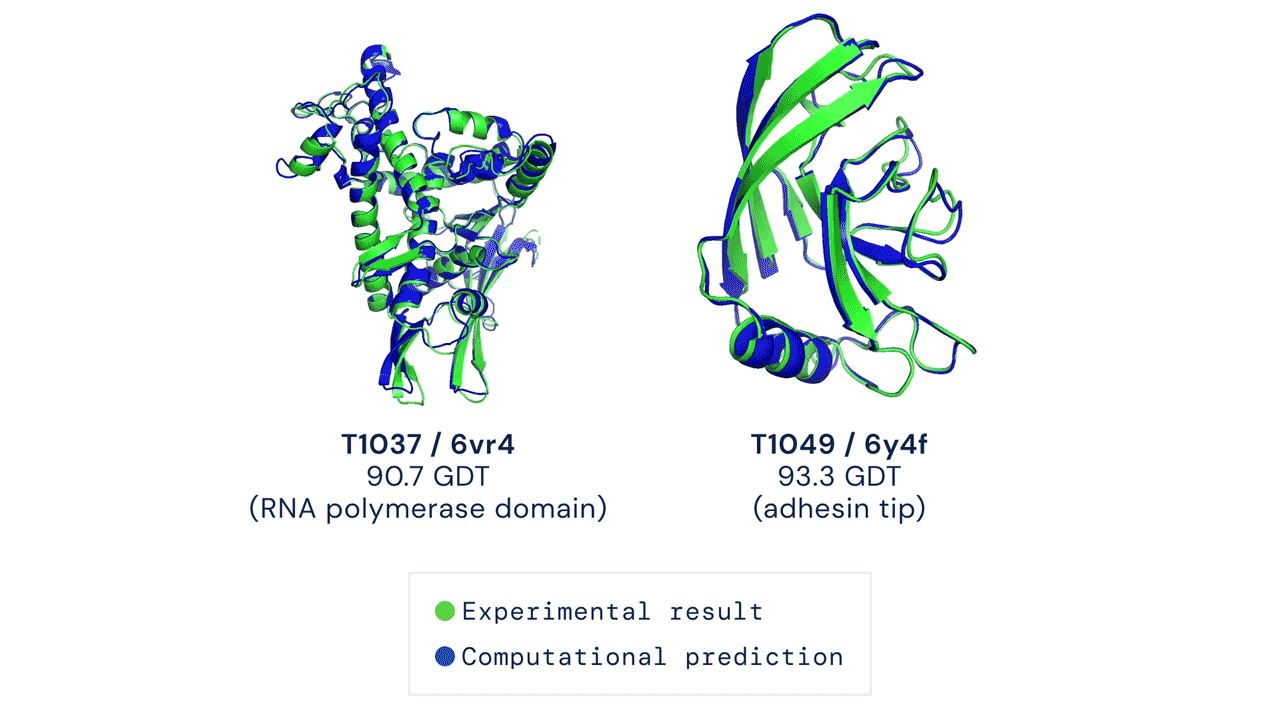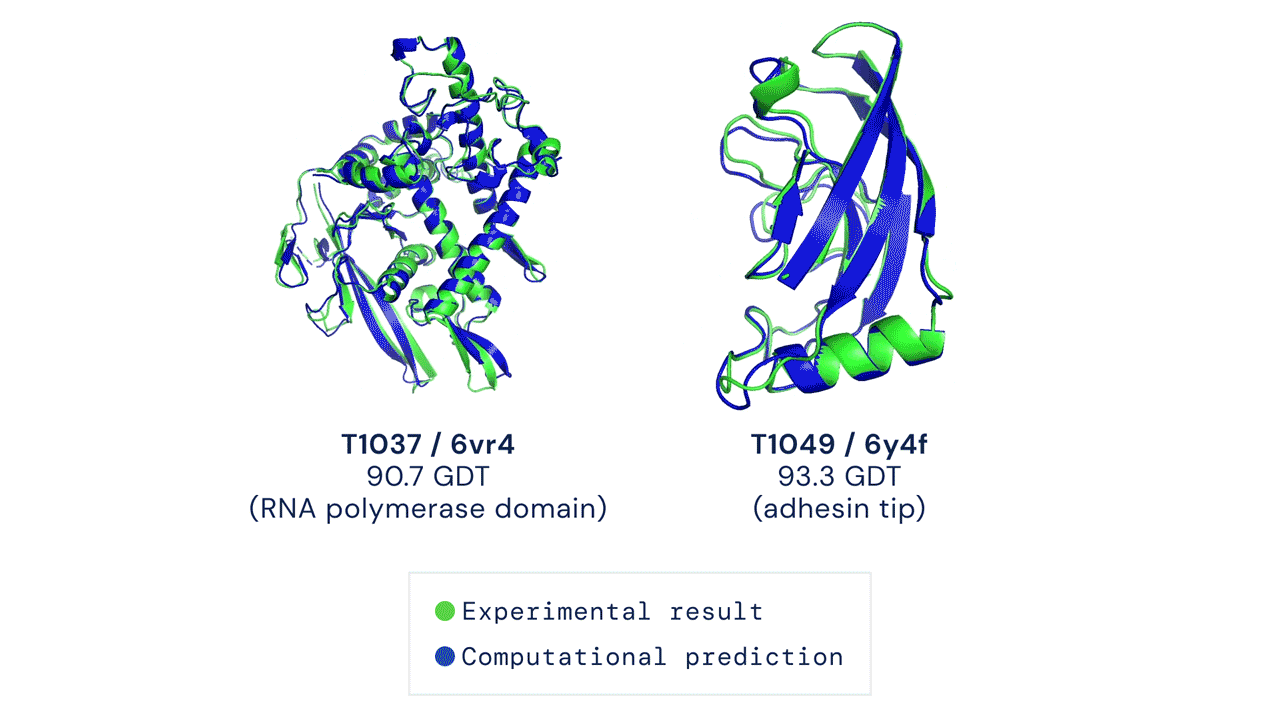
Joe Greener, a cambridge-i MRC laboratóriumban molekuláris szimulációkkal foglalkozó bioinformatikus decemberben a Twitteren fejtette ki a véleményét az AlphaFold2-ről, és kiemelte, hogy a módszer már most segít a kísérleti térszerkezet megállapításban, az a tény pedig, hogy az AlphaFold2-t nyílt forráskóddal közölték, sokat segített alkalmazhatóságán. A kísérleti térszerkezet-megállapítás támogatására jó példa Ioannis Skalidis és kollégáinak a strukturális biológiával foglalkozó Structure szakfolyóiratban idén január 28-án közölt tanulmánya. Ebben a molekuláris szerkezeteket feltáró kriogén-elektronmikroszkópos vizsgálatokat az AlphaFold2 segítségével végzett, atomi szintű modellezéssel kombinálják az anyagcserefolyamatokban résztvevő fehérjék szerkezetének vizsgálatához.
Az AlphaFold2 gyógyszerkutatásban való alkalmazási lehetőségeit pedig jól mutatja a gépi tanulással foglalkozó kutató, Russell Kaplan által szintén januárban megosztott történet, miszerint az AlphaFold2 egy barátja által vezetett biotechnológiai startupnál a korábbi egy hónapról néhány órára csökkentette a hatóanyag-jelölt fehérjék megtalálását, melyek megfelelő térszerkezetüknél fogva a várt terápiás hatást válthatják ki a célzott ráksejteknél.
Az Európai Bioinformatikai Intézet január 28-án 17, elhanyagolt trópusi betegségeket okozó élőlény, és 10, az antibiotikumok elleni rezisztencia szempontjából lényeges baktérium összesen 190 ezer fehérjéjének szerkezetét közölte. Ezzel az intézet AlphaFold2-adatbázisában megtalálható és mindenkinek elérhető fehérjeszerkezetek száma majdnem elérte az egymilliót.
De hogyan működik az AlphaFold2?
Az AlphaFold2 a 2018-as CASP13 versenyen résztvevő AlphaFold jelentősen továbbfejlesztett és átírt verziója, amely a két évvel későbbi CASP14-en sokkal jobbnak bizonyult minden más módszernél. Az AlphaFold2 a fehérje főlánc (peptidgerinc) szerkezet-meghatározásának átlagos pontosságát nézve egy szénatom méreténél is precízebbnek bizonyult: 0,96 angströmöt ért el, amivel majdnem háromszorosan túlszárnyalta a második legjobb módszert. Ha a fehérjék főláncát jól meghatározta, az AlphaFold2 képes az oldalláncok elhelyezkedését is pontosan megadni, és az összes atomra nézve 1,5 angström pontosságot elérni. A modell emellett aminosavanként megadja a létrehozott szerkezet pontosságát, ami a kutatók szerint az előrejelzés felhasználását segítheti.
A mély tanulási algoritmus alapjaiban épít a kísérletileg meghatározott, a Protein Data Bank (PDB) adatbázisba az elmúlt évtizedek során elhelyezett fehérje-térszerkezetekre, amelyeket bemeneti adatokként használ neurális hálóihoz. Az AlphaFold2 magukat a háromdimenziós struktúrákat, legalábbis azokban a nehezebb atomok helyzetét az aminosavsorrend segítségével, valamint azonos evolúciós eredetű fehérjék egymáshoz illesztett aminosavsorrendjének (multiple sequence alignment) figyelembevételével találja meg. Az AlphaFold2 mély tanuláshoz használt neurális hálózatának egyik fő alkotórésze az Evoformer nevű komponens, amely a rokon fehérjék egymáshoz illesztett aminosavsorrendjével a háromdimenziós szerkezet megoldását egy gráfproblémaként kezeli, illetve képes az ezen aminosavsorrendek együttese által tartalmazott fehérjeevolúciós információt feldolgozni. A neurális hálózat másik része ismétlési javítással dolgozik, amely a kutatók szerint nem sok extra tanulási időt igényel, viszont jelentősen növeli az AlphaFold2 pontosságát.
Az AlphaFold2 egyik fontos innovációja az, hogy a neurális hálózat a fehérjéken belül nem csak helyi, hanem nagyobb távolságú kölcsönhatásokkal is számol – vélekedik a Nature-ben tavaly megjelent véleménycikkében Mohammed AlQuraishi, a fehérjék térszerkezetének gépi tanulásos módszerekkel való meghatározásának szakértője. Mindezt a fehérjék geometriájáról meglévő alapvető ismeretek segítségével teszi, úgy, hogy közel 200-szor megismétli a lépést a végső háromdimenziós struktúra finomításához, anélkül, hogy ez elérhetetlen szintű számítási kapacitást igényelne.
Több tízezer emberi fehérje struktúrája lett meghatározva
Az emberi proteom 98,5 százalékának AlphaFold2 általi meghatározása jelentős eredmény, ugyanakkor fontos hozzátenni, hogy a fehérjéket alkotó aminosavakra vetítve csak 58 százalékban sikerült megbízható térszerkezeteket előállítani. Magukra a fehérjékre lefordítva ez azt jelenti, hogy 43,8 százalékuknál sikerült a szerkezet legalább háromnegyedét megbízhatóan feltárni. A magas megbízhatósághoz, ami aminosavakra vetítve csak 35,7 százalékban jött létre, arra volt szükség, hogy az AlphaFold2 a fehérjék főlánca mellett az oldalláncokat is pontos orientációban határozza meg.
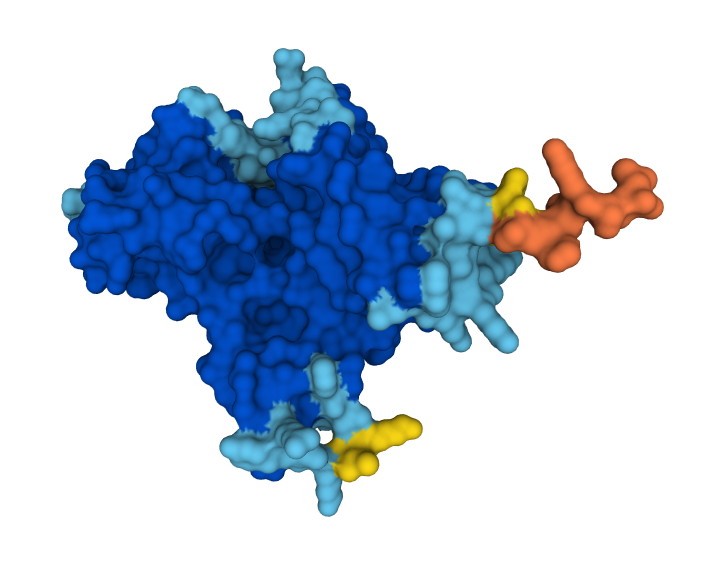
A kutatók néhány példát is hoznak tanulmányukban arra, hogyan világítanak meg molekuláris szintű folyamatokat az AlphaFold2 által megadott térszerkezetek. Ezek egyike a glükóz-6-foszfatáz enzim, ami a glükóz szintézisének utolsó lépését katalizálja, így kulcsfontosságú szerepet játszik a vércukorszint fenntartásában. A térszerkezetnek köszönhetően most először kiderült, hogy a fehérje kötőhelyén egy glutaminsavnak nevezett aminosav helyezkedik el, ami valószínűleg stabilizálja a kötőhelyet, és szabályozza az ahhoz való hozzáférést.
AlQuraishi véleménycikkében kifejtette azt is, hogy az AlphaFold2 által meghatározott térszerkezetek nem csak strukturális biológusoknak, hanem a fehérjék mozgását vizsgáló biofizikusoknak, vagy a fehérjék egymással való kölcsönhatását tanulmányozó molekuláris biológusoknak is hasznosak lehetnek, mint ahogy ez a glükóz-6-foszfatáz példáján látszik is.
Patrick Cramer szerint az AlphaFold2 és utódjai előtt álló következő kihívás a fehérjék és nukleinsavak (fehérje-RNS és fehérje-DNS komplexek) által közösen alkotott struktúrák meghatározása kell, hogy legyen, amelyekhez a fehérje-DNS együttesek esetén már elegendő adat áll rendelkezésre. AlQuraishi ezzel szemben a fehérjék nem túl jól definiált, „rendezetlen” térszerkezetű régióinak megoldására koncentrálna inkább, amelyek szerinte továbbra is alapvető problémát jelentenek az AlphaFold2-nek, és szerepük igencsak lényeges, mivel az emberi proteomban a fehérjéket alkotó aminosavak 37–50 százalékát érintik.
Kapcsolódó cikkek a Qubiten:
Fél évszázados problémát oldott meg, egy csapásra forradalmasíthatja a biológiát a DeepMind algoritmusa
A fehérjék 3D alakzatainak aminosav-szekvenciákból való hajszálpontos előrejelzésével a DeepMind szoftvere átalakítja mindazt, amit a biológiáról eddig tudtunk; évekkel felgyorsítja a sejtek működésének megértését, a fehérjetervezést, a gyógyszerkutatást és a hulladékot lebontó enzimek tervezését.

A mesterséges intelligencia végre feltárhatja a fehérjék szerkezetének rejtélyeit
Egyre közelebb kerülünk ahhoz, hogy pontosan meg tudjuk jósolni a fehérjék szerkezetét, ami forradalmasíthatja a biokémiát, és közelebb vihet súlyos betegségek gyógyításához is.