Soha nem látott fehérjéket álmodik meg a ProteinMPNN mesterséges intelligencia

A fehérjék térbeli szerkezetének feltárása után most új fehérjék tervezését is forradalmasíthatja a mesterséges intelligencia. A ProteinMPNN mélytanulási algoritmus sokkal gyorsabb a korábbi módszereknél, és nagyobb eséllyel generál működő molekulákat, ami megkönnyítheti a kutatóknak új, terápiákhoz szükséges fehérjék és nanostruktúrák létrehozását.
A Washingtoni Egyetem biokémiai tanszékének és fehérjetervezési intézetének kutatói két a Science-ben közölt tanulmányban számoltak be fejlesztésükről szeptember közepén. David Baker biokémikus szerint az élővilágban előforduló fehérjék jóval kevesebb mint 1 százalékát fedik le a lehetséges szerkezeteknek, az új szoftverek pedig régóta húzódó orvostudományi, energiatudományi, és technológiai problémákat oldhatnak meg.
Justas Dauparas és kollégái az új algoritmust és kísérleti tesztelését mutatták be, míg Basile Wicky és munkatársai ismertették, hogyan képes a ProteinMPNN azonos fehérje-alegységekből felépülő, változatos struktúrákat megálmodni. Ez a kutatók szerint megnyithatja az utat a mainál bonyolultabb, nanotechnológiás molekuláris gépek létrehozásához.
Két éve a DeepMind AlphaFold2 algoritmusa minden korábbi számítási módszernél pontosabban tárta fel, hogy a fehérjék aminosavsorrendje milyen funkcionális térszerkezetet eredményez. A fehérje-feltekeredést gyakorlati szemszögből szinte megoldó AlphFold2 azóta már 200 millió fehérje szerkezetét határozta meg, amik elérhetők egy publikus adatbázisban.
Az AlphaFold2-vel ellentétben a GitHub-ról letölthető ProteinMPNN új, a természetben nem előforduló fehérjék aminosav-sorrendjének tervezésére szolgál, amik szintézisük után feltekerednek, és kívánt szerkezetű molekulákká válhatnak. A biokémikusok erre eddig többnyire a Rosetta fehérjetervező szoftvert használták, aminél a ProteinMPNN az amerikai kutatók által kísérletileg igazoltan hatékonyabb, és közel 200-szor gyorsabb.
„A ProteinMPNN-hez hasonló mélytanulási módszerek átírták a játékszabályokat. Megrajzolod a fehérjédet, megnyomsz egy gombot, és kapsz valamit, ami tízből egyszer működik.” – mondta a Nature-nek Arne Elofsson, Stockholmi Egyetemen dolgozó bioinformatikus, aki nem vesz részt a kutatásban.
Több más kutatócsoport is fejleszt gépi tanuláson alapuló fehérjetervező algoritmusokat. Ilyen a ProtGPT2 is, melyet a németországi Bayreuth Egyetem kutatói, Noelia Ferruz és kollégái idén nyáron ismertettek a Nature Communications-ben.
Nemcsak gyorsabb a korábbi módszereknél, de többször is tervez valóban működő molekulákat
Az eddig széles körben alkalmazott Rosetta szoftver a fehérjetervezést olyan energia-optimalizáló problémaként kezeli, ahol egy adott háromdimenziós szerkezethez a legkisebb energiaszintű aminosav-kombinációt keresi meg, egy rendkívül számításigényes folyamat során.
A fehérjék egy vagy több polipeptidláncból épülnek fel, amelyek peptidkötéssel egymáshoz kapcsolódó aminosav-maradékok láncolatából állnak. A főlánc (peptidgerinc) a polipeptidláncnak a nagyrészt ismétlődő, a lánc változatosságáért felelős aminosav oldalláncoktól mentes része.
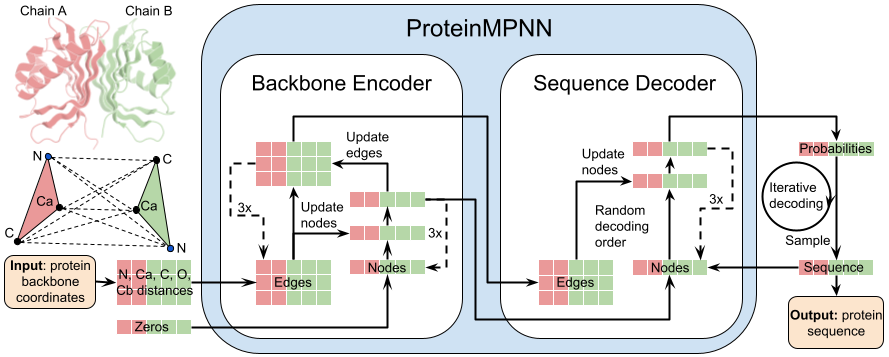
A ProteinMPNN a fehérje szekvenciáját a főlánc jellemzői, például az egyes szénatomok közti távolság vagy a szénatomok egymáshoz viszonyított orientációja alapján határozza meg. Dauparas és kollégái több ezer nagy részletességű, kísérleti módszerekkel meghatározott fehérjestruktúra segítségével tanították az algoritmust, ami kezdetben így 50 százalékos pontosságot ért el.
Az algoritmus pontosságát a kutatók azzal mérik, hogy mennyire képes rekonstruálni egy létező fehérje aminosavsorrendjét (sequence recovery) annak ismert, háromdimenziós térszerkezetéből, ami lényegében az AlphaFold2 feladatának fordítottja. Az optimalizációk után a ProteinMPNN a Rosetta 32,9 százalékánál jelentősen jobban, 52,4 százalékos pontossággal volt képes térszerkezetükből valódi fehérjék aminosavsorrendjét rekonstruálni. Mindezt 100 aminosavmaradékra és egy processzorra vetítve 1,2 másodperc alatt tette, szemben a Rosetta 258,8 másodpercével.
Ha egy kicsit engedtek a kutatók az algoritmus pontosságából azzal, hogy nagyobb szimulált hőmérsékleten vizsgálódott, sokkal változatosabb szekvenciákat kaptak. Ez szerintük a legtöbb alkalmazásnál előnyös, mivel így több szekvenciát lehet tesztelni, és nagyobb az esélye, hogy valamelyik működni fog.
Új fehérjeszerkezeteket álmodik az algoritmus
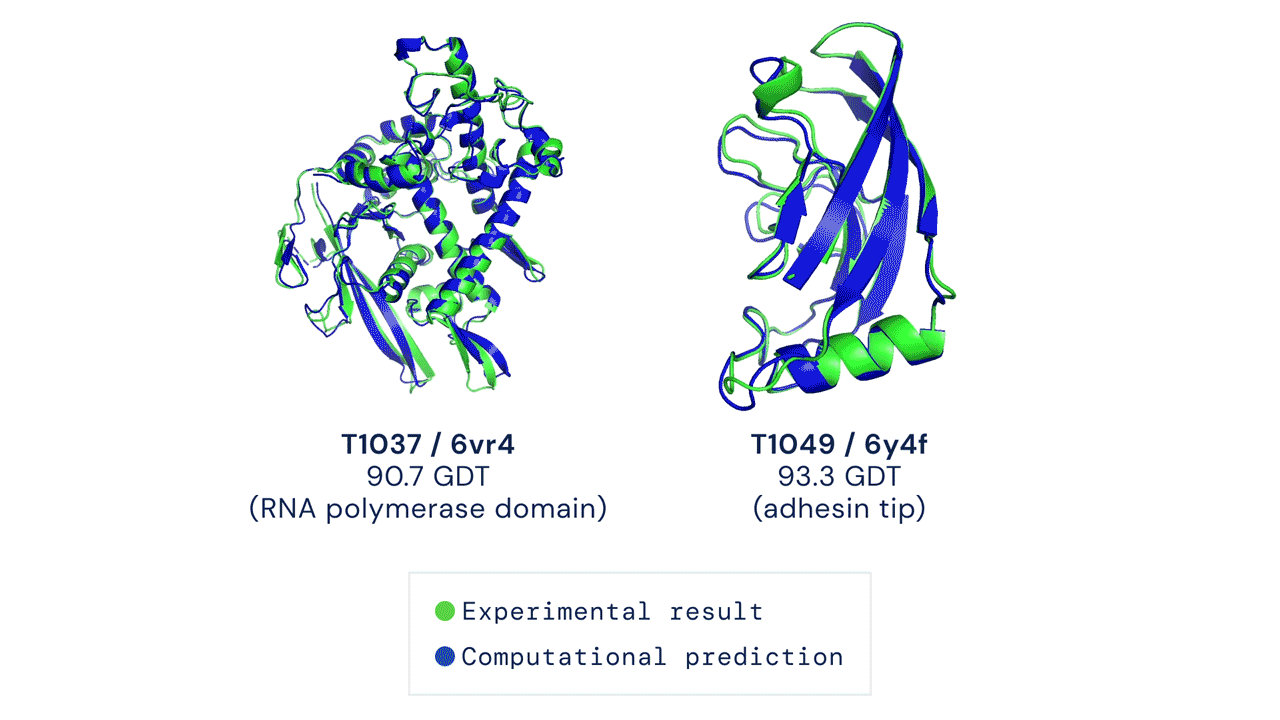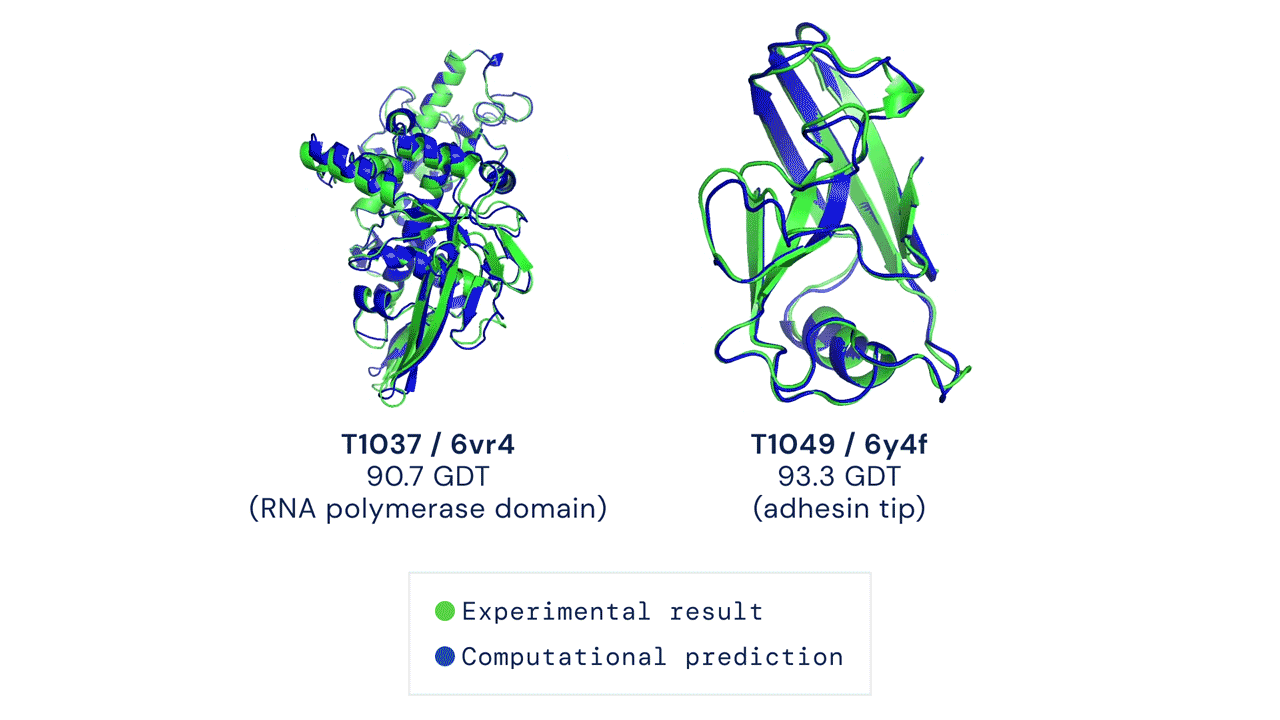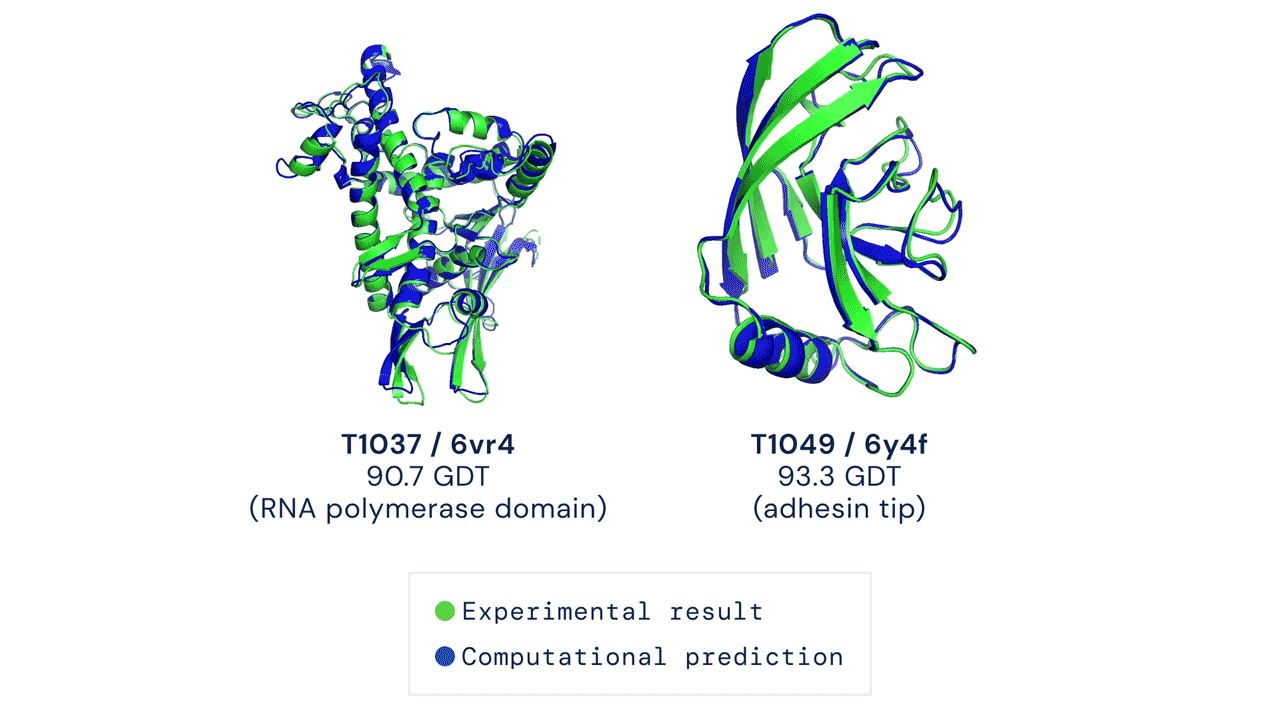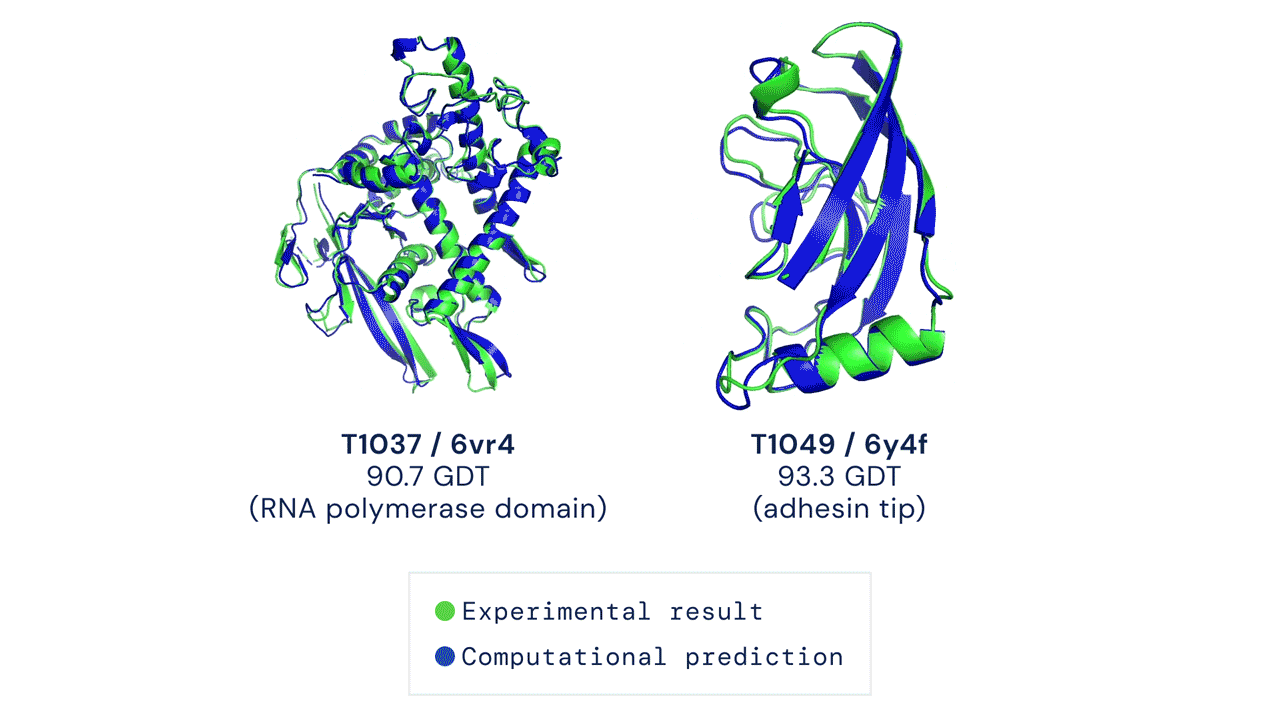
Mivel a rekonstrukciós precizitás a kutatók szerint nem vág egybe pontosan azzal, ahogyan a tervezett fehérjék valóban feltekerednek és funkcionálnak, a ProteinMPNN tudását kísérletileg is tesztelték. Egy ilyen kísérletben az algoritmus nanorészecskéket tervezett, amelyekhez hasonlót kísérleti oltásoknál is alkalmaznak. A 76 szekvenciatervet a kutatók Escherichia coli baktériumban szintetizálták, és 13 esetben kaptak molekuláris tömegük szempontjából megfelelő struktúrát. Az egyik térszerkezetét röntgendiffrakciós-szerkezetvizsgálati módszerrel meghatározták, ami nagyon jól egybevágott a kutatók által létrehozni kívánt fehérje modelljével.
Más kísérletek során a ProteinMPNN képes volt helyreállítani a Rosettával vagy AlphaFolddal készített hibás fehérjeterveket, így az algoritmus a Nature-nek nyilatkozó szakértő szerint egyfajta ellenőrzésként is használható lehet majd AlphaFold által gyártott szekvenciákhoz. Emellett a ProteinMPNN olyan bonyolult fehérjetervezési problémákkal is meg tudott küzdeni, amelyekkel a Rosetta nem. További előnye az algoritmusnak a kutatók szerint, hogy nincs szüksége arra, hogy adott fehérjetervezési problémához testre szabják, így többeknek elérhetővé teszi a fehérjetervezést.
„Azt találtuk, hogy a ProteinMPNN-el készített fehérjéknél nagyobb esély volt arra, hogy megfelelően felterednek, és így nagyon komplex fehérjeszerkezeteket is létre tudtunk hozni” – mondta Wicky. A kutató és kollégái a mélytanulási algoritmussal a természetes fehérjestruktúrákon túli lehetőségeket kívánták felfedezni, amihez egy hálózati hallucinációnak nevezett módszert alkalmaztak.
Ennek segítségével különböző szimmetrikus fehérjeszerkezeteket generáltak a ProteinMPNN-el, melyek azonos alegységből épülnek fel (homo-oligomer fehérje). Miután szintetizálták a struktúrákat, kriogén-elektronmikroszkópos (cryo-EM) vizsgálattal meghatározták a szerkezetüket. Kiderült, hogy a 192-ből 7 kristályszerkezete nagyon pontosan egyezett a számítógépes modellekkel. A kutatók szerint ez egy sokkal jobb arány, mint amit a Rosetta tipikusan el tud érni újonnan létrehozott (de novo) fehérjék esetén.
A szakemberek szerint mindez azt mutatja, hogy innentől a mélytanulási modelleknek köszönhetően a természetben előforduló fehérjék szerkezeti adatbázisain túlmutató, új fehérjevilág tárulhat fel. Ma még a nagyobb és komplexebb struktúrák megálmodásához nagy számítási kapacitásra van szükség, így Wickyék szerint a következő cél kevesebb paraméterrel rendelkező, könnyebben lefutó módszerek létrehozása lesz.
A címlapi kép forrása: Baker Lab, Institute for Protein Design, University of Washington.
* * *
Forró őszünk van, de vajon hogyan oldható meg a pedagóguskrízis? Hova vezet az erőltetett iskolai központosítás? Tényleg egyfajta kasztrendszerszer felé tart az ország? Októberi tudományos estünkre a magyar közoktatás problémáival készülünk: öt okos kutatóval, négy előadással, vitával és kerekasztallal, zárásként pedig improvizatív zenével és bárral. Mindez izgalmas helyszínen, a margitszigeti Kristályban vár majd rátok október 4-én, kedden 18:00-tól. Jegyeket itt tudtok venni, gyertek el, jó lesz!

Kapcsolódó cikkek a Qubiten:
Áttörést hoz a biológiába a minden eddiginél pontosabb fehérjekutató algoritmus, az AlphaFold2
A Google-féle DeepMind legújabb AI-modellje az aminosavak sorrendjéből egész pontosan megfejti a fehérjék háromdimenziós térszerkezetét. A mesterséges intelligencia forradalmasíthatja a gyógyszerkutatást: van olyan rákkutató cég, ahol az AlphaFold2 a korábbi egy hónapról néhány órára csökkentette a hatóanyag-jelölt fehérjék megtalálását.

A műanyagevő baktériumok már a küszöbön állnak, csak fel ne falják az egész konyhát
A PET-palackokat, csomagolóanyagokat, pólókat lebontani képes baktériumok és enzimek a laborban már jól teljesítenek, de kérdés, hogy ipari mértékben skálázhatók-e, és elég biztonságos-e ahhoz, hogy az éhes kis mikroorganizmusokat kiengedjük a kontrollált bioreaktorokból.

Forradalmi áttörés: a mesterséges intelligencia a DNS-ből megmondja, hogyan fognak kifejeződni a gének
Német kutatók olyan mélytanulási rendszert hoztak létre, amely több millió mérésen edződve képes pontosan előre jelezni, milyen gyakorlati hatással lesz egy adott szabályozó szekvenciának a gének átíródására – így az emberi megbetegedésekkel összefüggő régiók genetikai variánsait is feltárhatja.