A DeepMind új algoritmusa megjósolja, milyen betegségekhez vezethetnek a fehérjék hibái

Egy új algoritmus képes előre jelezni, hogy okozhatnak-e betegséget emberi fehérjéket módosító genetikai változások – állítják a Google anyacége (Alphabet) alá tartozó DeepMind kutatói. A kedden bemutatott AlphaMissense a mesterséges intelligencia (AI) segítségével előbbre viheti a genomikai információk egészségügyi felhasználását, de független szakemberek szerint hatása várhatóan nem lesz forradalmi.
A Jun Cheng és kollégái által a Science folyóiratban ismertetett algoritmus a cég áttörő fehérjekutató algoritmusára, az AlphaFoldra épül. Utóbbi 2020-ban, a CASP14 algoritmusversenyen forgatta fel a területet, miután nagy pontossággal képes meghatározni a fehérjék térbeli szerkezetét az őket felépítő aminosavak szekvenciájából, amivel a gyakorlatban megoldotta a molekuláris biológia egyik legnagyobb problémáját.
A DeepMind szakemberei azt állítják, az AlphaMissense előrejelzéseivel hamarabb megértjük majd, milyen funkcionális hatásai vannak azoknak az úgynevezett missense genetikai mutációknak, amelyek miatt egyetlen aminosav kicserélődik egy fehérjében. A missense variánsokból eddig négymilliót figyeltek meg, viszont mindössze két százalékukat osztályozták aszerint, hogy okozhatnak-e betegségeket (tehát patogének) vagy ártalmatlannak tűnnek.
A szerzők az AlphaMissense-szel most több tízmillió ilyen emberi variáns hatását jelzik előre, ami nagyságrendekkel növeli a meglévő predikciók számát. Emellett úgy vélik, algoritmusuk hosszabb távon segíthet megbetegedéseket okozó gének felderítésében, és javíthat a ritka genetikai megbetegedések diagnosztizálási arányain.
A Nature-nek nyilatkozó Arne Eloffson bioinformatikus szerint az AlphaMissense fejlettebb az eddigi, a mutációk hatását előrejelző megoldásokhoz képest, de „nem jelent hatalmas előrelépést”. Hasonlóan vélekedik Joseph Marsh, az Edinburgh-i MRC Humángenetikai Intézet bioinformatikusa is, aki szerint „valószínűleg ez most a legjobb előrejelző. De ez lesz a legjobb két vagy három év múlva is? Jó esély van arra, hogy nem”.
Hogy működik az AlphaMissense?
Az AlphaFoldhoz hasonló felépítésű új algoritmust emberi és főemlősökből származó adatokkal tanították, amik ezeknek a fehérjemutációkat okozó variánsoknak a populáción belüli elterjedtségét tartalmazzák. A kutatók egy aminosav-szekvenciát adnak bemenetként az algoritmusnak, amiből az utána kiadja, milyen hatása lehet betegségek kialakulásra az összes lehetséges, egy adott fehérjében bekövetkező egyetlen aminosavnyi változásnak. Az AlphaMissense annyiban tér el az AlphaFoldtól, hogy nem határozza meg, milyen következményei vannak ezeknek a mutációknak a fehérjék térszerkezetére.
Az algoritmus tanítása két fázisban történik. Az első az AlphaFoldhoz hasonló, ahol a szekvencia alapján egy fehérjelánc térszerkezetét próbálja meghatározni. Emellett megpróbálja azt is megjósolni, hogy melyik, a kutatók által „kitakart” aminosav található egy random pozícióban a tanuláshoz használt adatokban. Ezek az úgynevezett egymáshoz illesztett aminosavsorrendek (multiple sequence alignments), amikből az AlphaFold és az AlphaMissense evolúciós információt tud kinyerni, és így például a fehérje térszerkezetére következtetni.
A következőkben az AlphaMissense-t emberi fehérjék szekvenciáin pontosítják, valamint a már említett emberi és főemlős adatokon tanítják tovább. Ezeknél azokat a missense genetikai variánsokat, amik gyakran előfordulnak az emberi vagy főemlős populációkban, ártalmatlanként címkézik fel, a populációkból hiányzó variánsokat pedig patogénként.
A DeepMind kutatói azt állítják, az AlphaMissense számos, a számítógépes prediktorok teljesítményének összehasonlítására alkalmas benchmarkban lekörözi versenytársait, és az eddigi módszereknél nagyobb megbízhatósággal osztályozza a variánsokat. Továbbá azt találták, hogy az AlphaMissense az eddig jónak számító EVE algoritmushoz képest kiterjeszti a 90 százalékos biztonsággal meghatározott variánsok számát, 67-ről 93 százalékra.
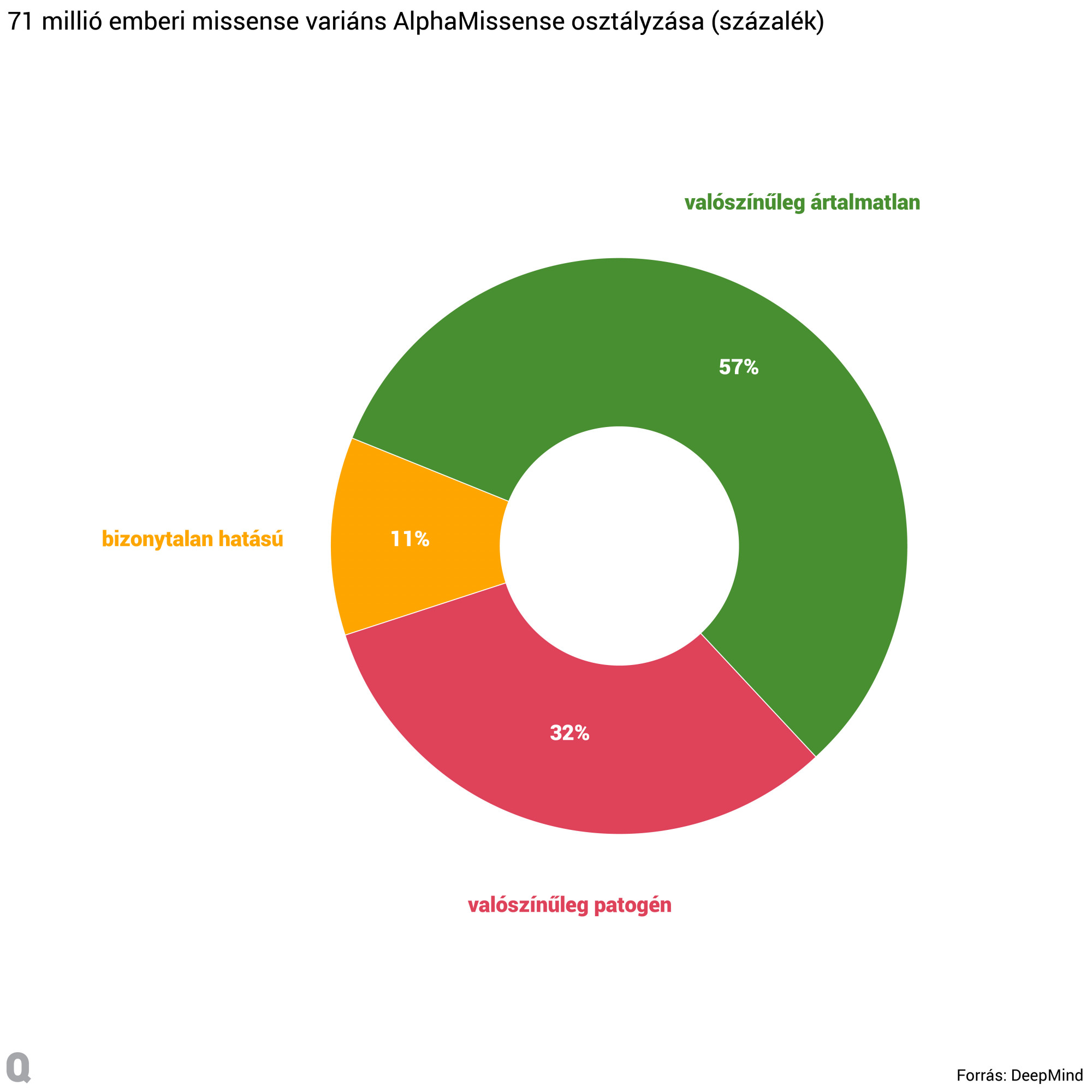
A DeepMind kutatói négy adatbázist és adatterméket bocsátottak a tudományos közösség rendelkezésére. Ezek többek közt 71 millió missense variánsra vonatkozó előrejelzést tartalmaznak, ami teljesen lefedi az emberi fehérjekészletet, a proteomot. Ennek a 71 millió variánsnak a többségét, 40,9 milliót „valószínűleg ártalmatlannak”, 22,8 milliót pedig „valószínűleg patogénnek” osztályozott az algoritmus. Emellett egy génszintű előrejelzést is kiadtak, ami azt adja meg, hogy egy gén összes lehetséges missense variánsa átlagosan milyen mértékben okozhat megbetegedést.
Nem ájult el tőle mindenki
Az AlphaMissense-t bemutató tanulmányt a Science-nek kommentáló Marsh és a Cambridge-i Wellcome Sanger Intézet genetikusa, Sarah Teichmann azt állítja, hogy jelenleg ezeket a számítógépes előrejelzéseket minimális mértékben lehet használni a genetikai diagnosztikában, legalábbis az amerikai orvosgenetikai és genomikai társaság (ACMG) ajánlása alapján. Bár Cheng és munkatásainak megközelítését kétségtelenül hasznosnak látják a variánsok hatásainak megállapításában és annak kiválasztásában, hogy melyekre érdemes nagyobb figyelmet fordítani, fontosnak tartják, hogy a „valószínűleg patogén” vagy „valószínűleg ártalmatlan” besorolásokat nem szabad összekeverni a több különböző bizonyítékon nyugvó klinikai definíciókkal.
A korábban Twitterként ismert X-en több kutató kifogásolta, hogy a szerzők nem tették közzé az eredmények reprodukálhatóságához szükséges összes technikai információt, például a modellhez tartozó súlyozásokat. A DeepMind szakemberei ezt a kutatási áttörések biztonságos és felelősségteljes publikálása iránti kötelezettségvállalással, valamint a potenciálisan nem biztonságos alkalmazások megakadályozásával indokolták. Ezt a magyarázatot meglehetősen szkeptikusan fogadta többek közt Mohammed AlQuraisihi is, aki a fehérjekutató algoritmusok egyik vezető szakértője.
Kapcsolódó cikkek a Qubiten:
Elképesztően fejlett fehérjekutató algoritmusok versengenek egymással, hogy feltérképezzék a legalapvetőbb biológiai folyamatokat
A fehérjék szekvenciájából 3D szerkezeteket előállító AlphaFold2 mesterséges intelligencia 2020-ban megoldotta a molekuláris biológia egyik legnagyobb problémáját. Az algoritmusok legújabb versenyén az is körvonalazódott, hogy merre halad tovább a terület.
A Meta forradalmi algoritmusa feltárta a fehérjeuniverzum sötét anyagát
A Facebook anyavállalatának mesterséges intelligenciával foglalkozó kutatói az eddigi módszereknél sokkal gyorsabb fehérjekutató algoritmust hoztak létre, amely egy csapásra 600 millió fehérje háromdimenziós szerkezetét fejtette meg.
200 millió fehérje térszerkezetét tárta fel egyetlen év alatt a DeepMind fehérjekutató algoritmusa
Az embereknek több évtized alatt 100 ezret sikerült.
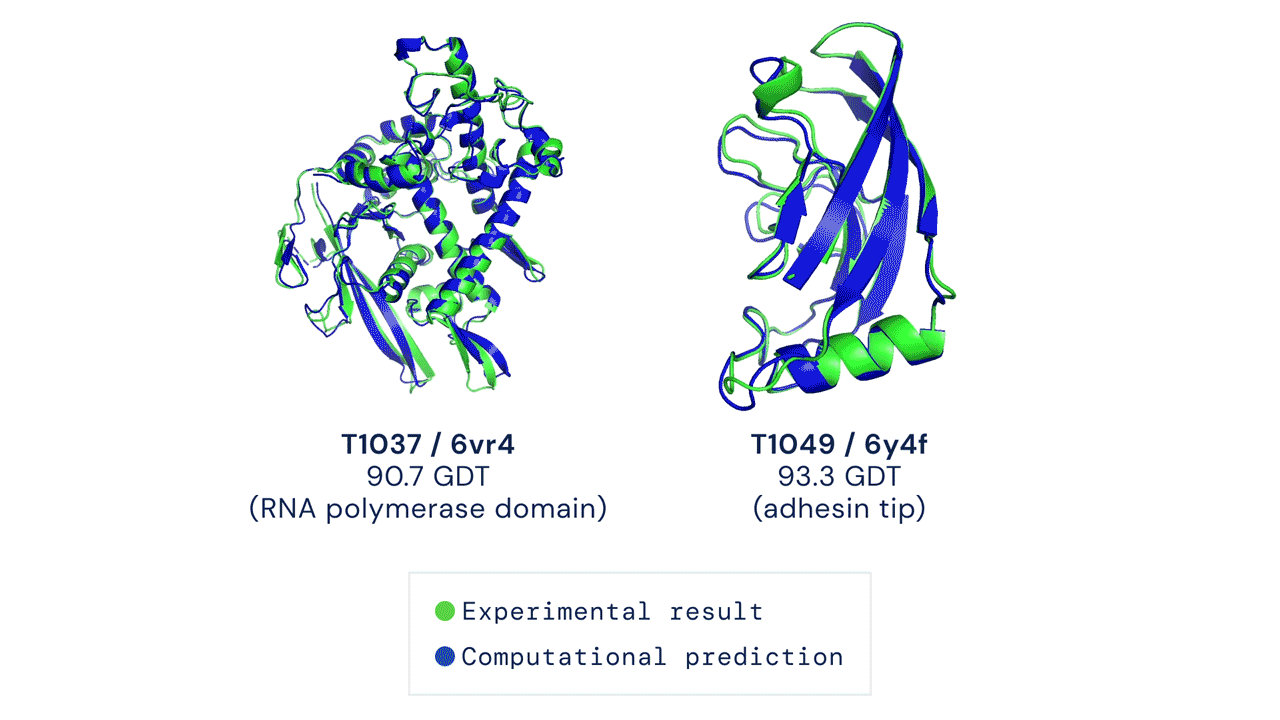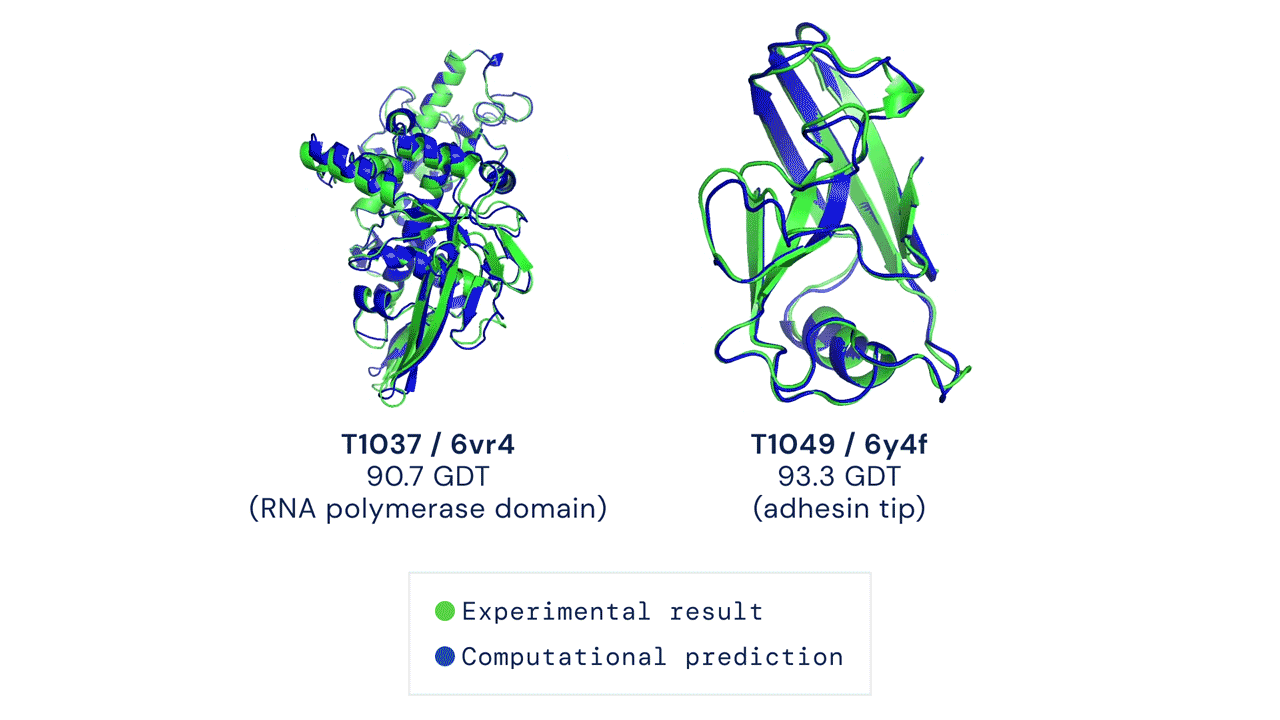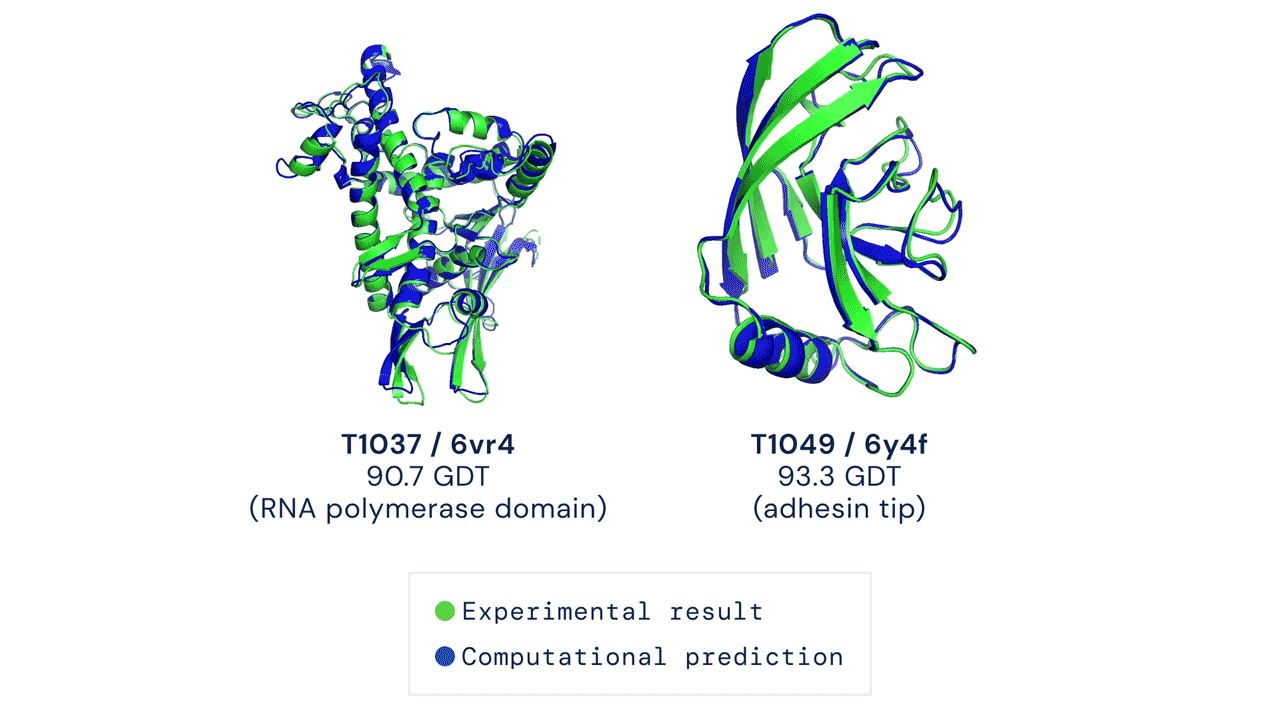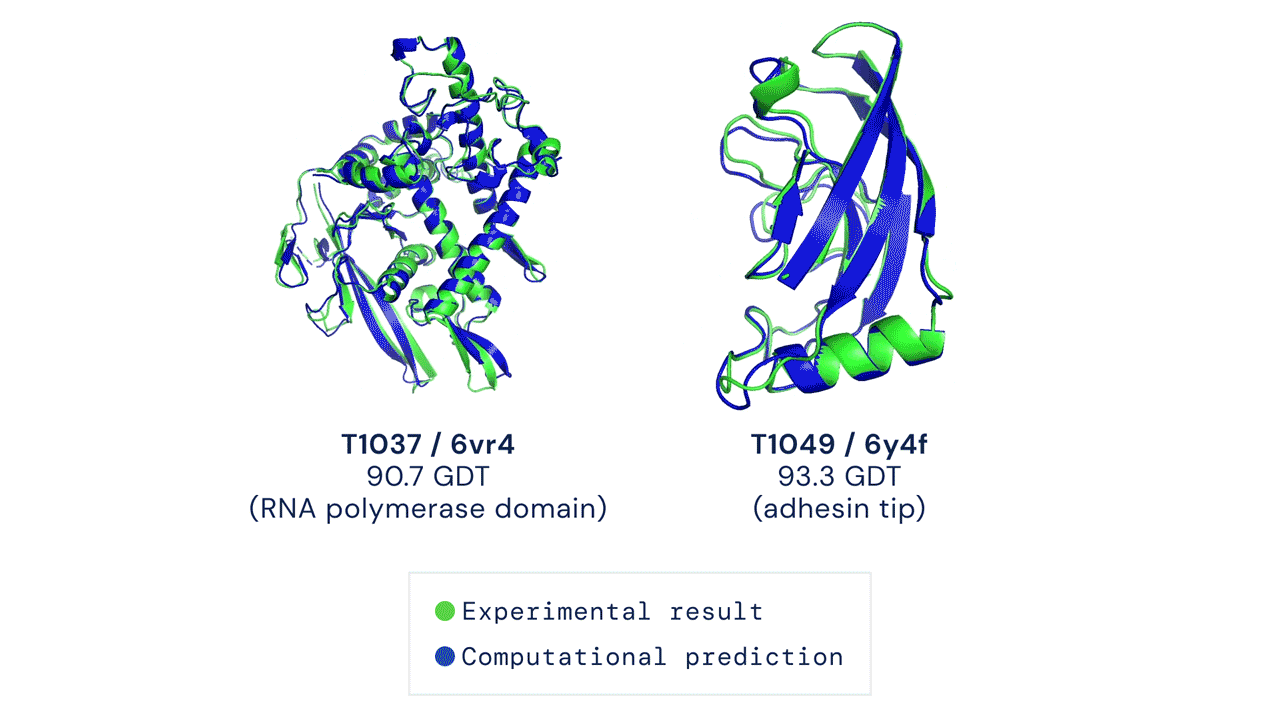
Áttörést hoz a biológiába a minden eddiginél pontosabb fehérjekutató algoritmus, az AlphaFold2
A Google-féle DeepMind legújabb AI-modellje az aminosavak sorrendjéből egész pontosan megfejti a fehérjék háromdimenziós térszerkezetét. A mesterséges intelligencia forradalmasíthatja a gyógyszerkutatást: van olyan rákkutató cég, ahol az AlphaFold2 a korábbi egy hónapról néhány órára csökkentette a hatóanyag-jelölt fehérjék megtalálását.