A Meta forradalmi algoritmusa feltárta a fehérjeuniverzum sötét anyagát

A Facebook anyavállalata, a Meta már a fehérjekutató algoritmusok területén is versenyez a legnagyobb AI-cégekkel, és most rendkívüli eredményekről számolt be: ESMFold nevű mesterséges intelligenciája több mint 600 millió fehérje háromdimenziós szerkezetét fejtette meg. Az új módszer példátlan betekintést ad a fehérjeszerkezetek valódi változatosságába, ami gyógyszerkutatási és biotechnológiai alkalmazásokkal kecsegtet – derül ki az október végén közzétett preprint (szakmai lektoráláson még át nem esett) tanulmányból.
A Lin Cö-ming, a New York-i Egyetem és a Meta AI csoportjának kutatója és kollégái által kifejlesztett, ESM-2 nyelvi modelleken alapuló ESMFold az eddigi megoldásoknál több mint 10-szer gyorsabb lehet, és az esetek többségében hasonlóan precíz eredményt ad, mint a legjobb fehérjekutató algoritmus, a DeepMind-féle AlphaFold2, amely eddig 200 millió fehérje szerkezetét tárta fel.
A mostani áttörés jelentőségét elsősorban az adja, hogy az algoritmust olyan fehérjék százmillióira szabadították rá, amelyekről eddig szekvenciájukon kívül nagyon keveset tudtunk. Az elmúlt években lehetővé vált, hogy kutatók különböző környezetekből – tengeraljzatoktól az emberi mikrobiomon át egészen régészeti lelőhelyekig – vett mintákban feltárják az összes ott megtalálható élőlény genetikai információját. A DNS-bázissorrend meghatározásával a metagenomok szekvenálása lehetővé teszi a gének által kódolt fehérjék szekvenciájának megismerését is. A fehérjék, amelyeket peptidkötésekkel egymáshoz kapcsolódó aminosav-maradékok láncolatából álló polipeptidek építenek fel, központi szerepet játszanak az élő szervezetek működtetésében.
Ahogy a metagenomok feltárták a mikroorganizmusok sokáig rejtve maradt változatosságát, az ESMFold most ugyanezt ismételheti meg a fehérjék világában. Eugene Koonin neves orosz-amerikai evolúciógenetikus szerint „a metagenomika a fehérjék hatalmas változatosságáról lebbenti fel a fátylat, amik közül eddig jó néhány, vagy akár a többségük is ismeretlen volt a tudománynak, és amik potenciálisan a biológiai életet érintő legalapvetőbb kérdésekre adhatnak választ”.
A kutató szerint „számos új, metagenomikával felfedezett fehérje a fehérjeuniverzum sötét anyagát alkotja, mivel a struktúrájuk és biológiai szerepük ismeretlen. Jelentős előrelépés ilyen mennyiségben ezeket a struktúrákat feltérképezni, ami példátlan betekintést ad majd a fehérjeszerkezetek és funkciók világába”.
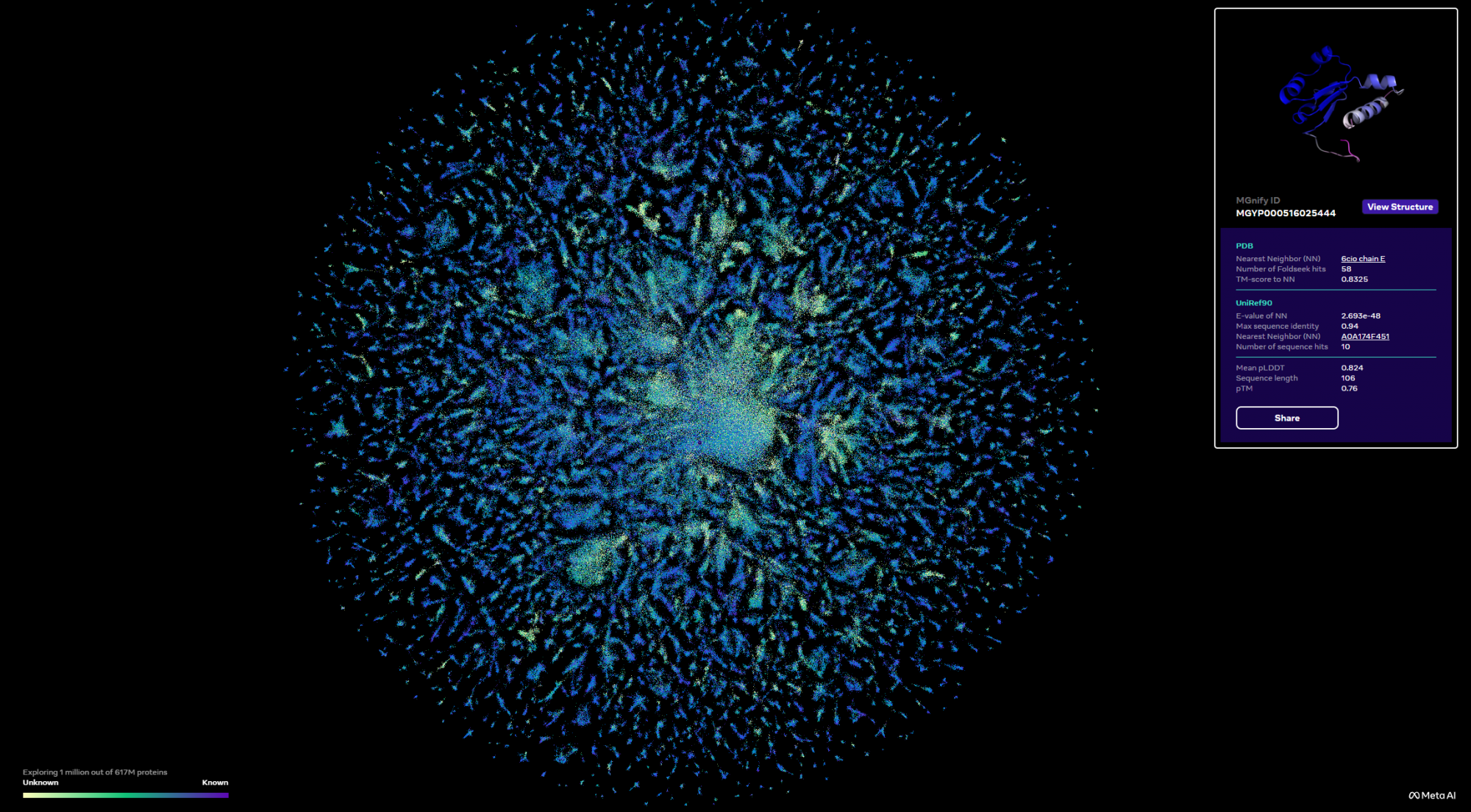
A kutatók az algoritmus teszteléséhez a MGnify90 nevű metagenomikai adatbázist használták, az annak 99 százalékát felölelő 617 millió fehérje szekvenciájához tartozó térszerkezet meghatározásával. Az így létrehozott ESM metagenomikai atlasz, valamint a nyelvi modellek bárkinek nyíltan elérhetők GitHubon.
Hogyan működik az ESMFold?
Az ESM-2 nyelvimodell-családot a kutatók 8 millióról 15 milliárd paraméterre bővítették ki, és a korábbi ESM-1b-hez képest több területen javítottak rajta. Az ESM-2 modellek a tanulás során előre jelzik a véletlenszerűen kiválasztott aminosavak típusát egy fehérjeszekvenciában, annak megfigyelése alapján, hogy hol helyezkednek el egy szekvencián belül. Ezzel a modell a kutatók szerint megtanulja, hogyan függenek egymástól a szekvenciában az egyes aminosavak, és miként alakul ez a mintázat a fehérjék evolúciójában.
A tanuláshoz 65 millió egyedi fehérjeszekvenciát használtak a kutatók, és ahogy a paraméterszámot lényegesen kibővítették, jelentős javulást figyeltek meg a fehérjemodellekben. Úgy vélik, hogy mivel az ESM-2 csak szekvenciákon tanul, az információ, amit a fehérjeszerkezetről megalkot, a szekvenciákban fellelhető evolúciós mintázatok felderítésén alapszik. A nyelvi modellek a kutatók szerint megtanulhatják azt, hogy a fehérjeszekvenciák milyen biológiai tulajdonságai alakítják ki ezeket a mintázatokat.
Az ESM-2 modelleken alapuló ESMFoldnak csak a fehérjeszekvenciát kell megadni, és eredményül megkapjuk az atomi felbontású struktúrát, annak előre jelzett bizonytalanságával. Az ESMFold a szerzők szerint sokkal egyszerűbb, mint más fehérjekutató algoritmusok, amelyek mélyen integrálják működésükbe az evolúciósan rokon, egymáshoz illesztett fehérjeszekvenciákban rejlő információt. Az ESMFoldnak nincs szüksége ezekre a számításigényes többszörös szekvenciaillesztésekre (multiple sequence alignment), ami részben magyarázza, hogy miért fejezi be a munkát hamarabb más megoldásoknál.
A gyakorlatban ez azt jelenti, hogy az NVIDIA gépi tanulásra optimalizált V100 grafikus kártyáján az ESMFold egy 384 aminosav-maradékból felépülő fehérje szerkezetét 14 másodperc alatt fejti meg. Ezzel 6-szor gyorsabb, mint az AlphaFold2, és a különbség akár 60-szorosra nőhet rövidebb szekvenciák esetén. Könnyen belátható, hogy ezek a különbségek egy több millió szekvenciát tartalmazó adatbázis esetén gyorsan nagyon jelentősre nőnek.
Tényleg jobb, mint az AlphaFold2?
A kutatók szerint az ESMFold az adatbázisban található fehérjék több mint felén hasonló pontosságot ér el, mint a fehérjefeltekeredés problémáját praktikusan megoldó AlphaFold2 – ez még néhány nagyobb fehérje, például az 540 aminosav-maradékból felépülő T1076 membránfehérje esetén is igaz. Az is kiderült, hogy az ESMFold és az AlphaFold2 között nem nagyon tér el, hogy a fehérjeszerkezetek mely részei pontatlanabbak. A szakemberek úgy látják, ez annak a jele, hogy a nyelvi modell lényegében az AlphaFold2 működésének egyik alapját adó többszörös szekvenciaillesztések nélkül is képes az abban rejlő információt megtanulni.
A 617 millió fehérje-térszerkezet meghatározása 2 hétbe telt egy 2000 grafikus kártyát tartalmazó számítási klaszternek. Az eredményként kapott fehérjestruktúrák közel 36 százaléka (225 millió) lett magas megbízhatóságú, és 18 százaléka nagyon magas megbízhatóságú, ami a kutatók szerint már összevethető a kísérleti úton mért fehérjeszerkezetek precizitásával. Az eredmények különlegességét talán az jelzi legjobban, hogy több tízmillió előre jelzett térszerkezet egyetlen egyezést sem mutat az eddig kísérletileg meghatározott struktúrákkal, vagyis ezek teljesen újak a biológusoknak.
Burkhard Rost, a Müncheni Műszaki Egyetem bioinformatikusa a Nature-nek elmondta, hogy a Meta nyelvi modelljének gyorsasága és pontossága lenyűgöző, de kérdéses, hogy valóban előnyősebb-e az AlphaFold precíziójához képest, amikor a fehérjék szerkezetének metagenomikai adatbázisokból történő meghatározásáról van szó. A kutató szerint a nyelvi modellek igazán annak megállapítására jók, hogyan változtatják meg mutációk a fehérjék szerkezetét.
A szakemberek szerint a gyors és pontos fehérjeszerkezet-előrejelzés elhozhatja azt a korszakot, amikor minden olyan fehérje térszerkezetét megismerhetjük, amelynek már ismert a szekvenciája. Úgy vélik, hogy a metagenomikai atlaszban a nagyon magas megbízhatóságú előrejelzések a gyakorlatban is alkalmazhatók lehetnek, például ha kutatók aktív helyek működését szeretnék megérteni. Ezek olyan fontos régiók az enzimeken, ahová úgynevezett szubsztrátmolekulák kötnek, és ahol a kémiai reakciók katalízise lezajlik.
A Meta kutatói azzal zárják tanulmányukat, hogy a jelenlegi modelljeik még nagyon messze vannak az algoritmus fejlettségének, a szekvenciaadatoknak és a számítási kapacitásnak az elméleti határaitól, így optimisták azt illetően, hogy a további fejlesztés még pontosabb fehérje-térszerkezeti előrejelzéshez vezethet.
Kapcsolódó cikkek a Qubiten:

Soha nem látott fehérjéket álmodik meg a ProteinMPNN mesterséges intelligencia
Az új mélytanulási algoritmus forradalmasíthatja a terápiákhoz és nanotechnológiás szerkezetekhez szükséges fehérjék tervezését. A ProteinMPNN sokkal gyorsabb, mint a korábbi eszközök, és az általa tervezett molekulák többnyire a valóságban is működnek.
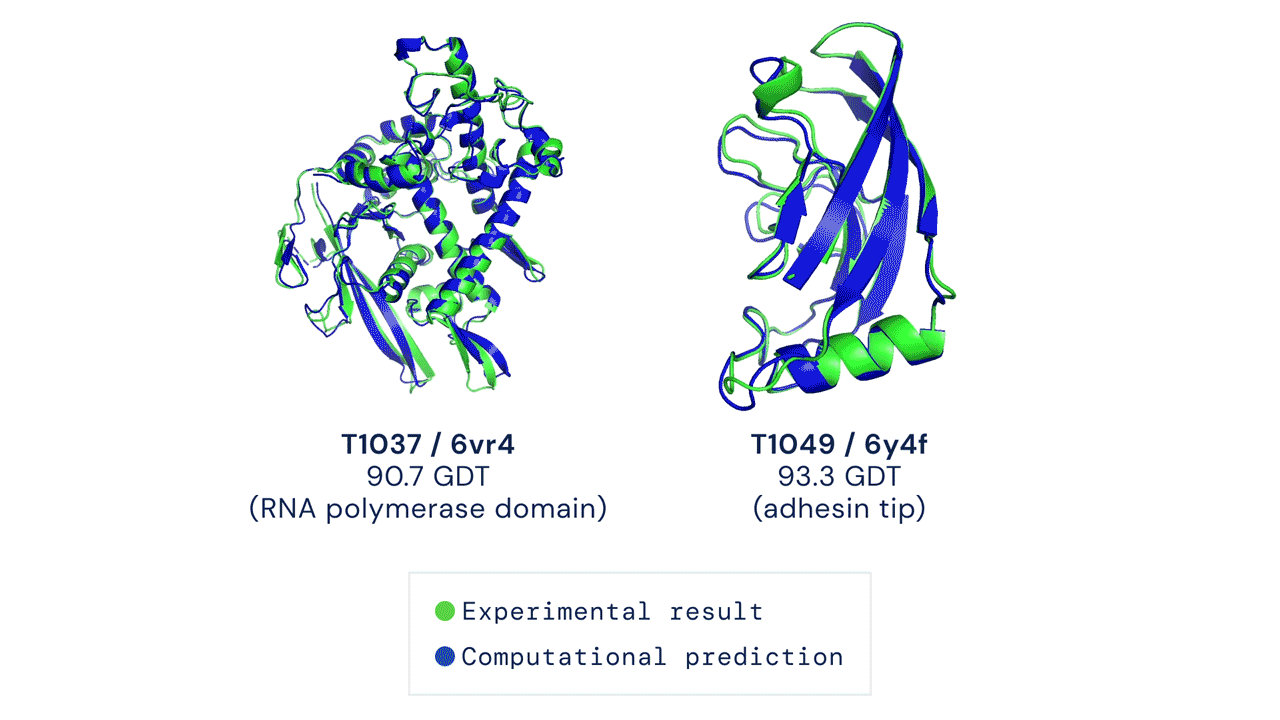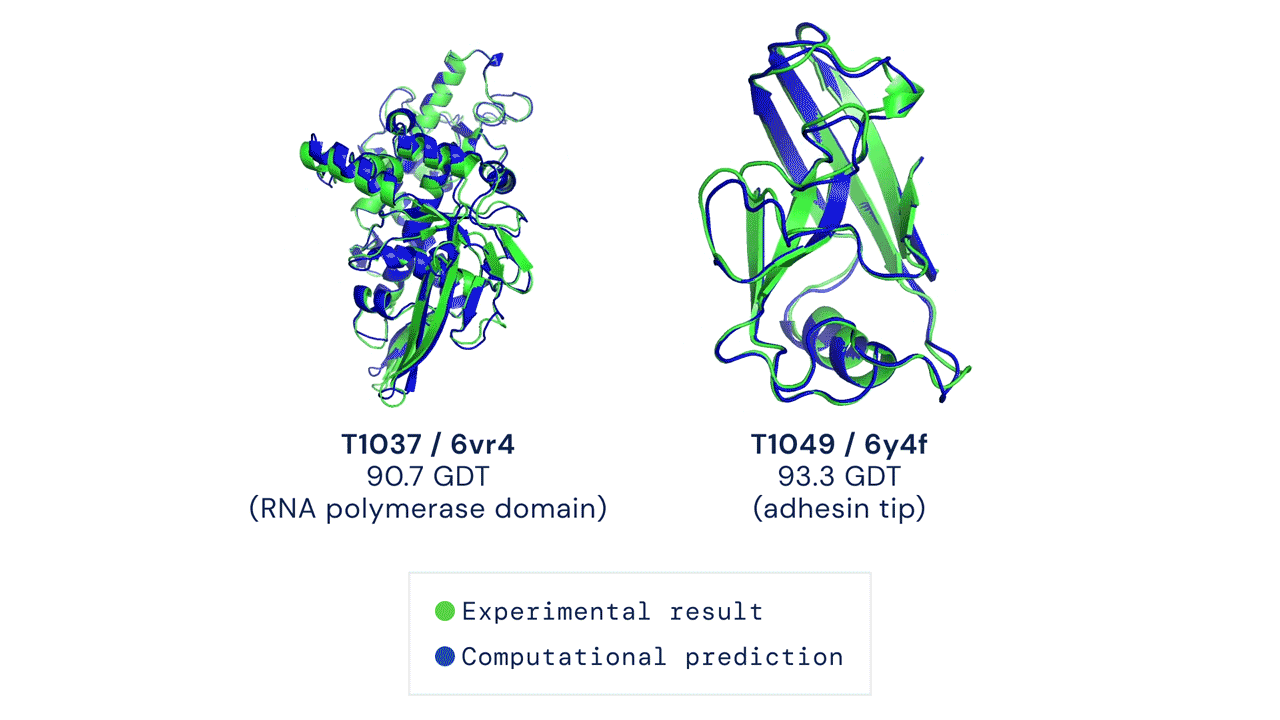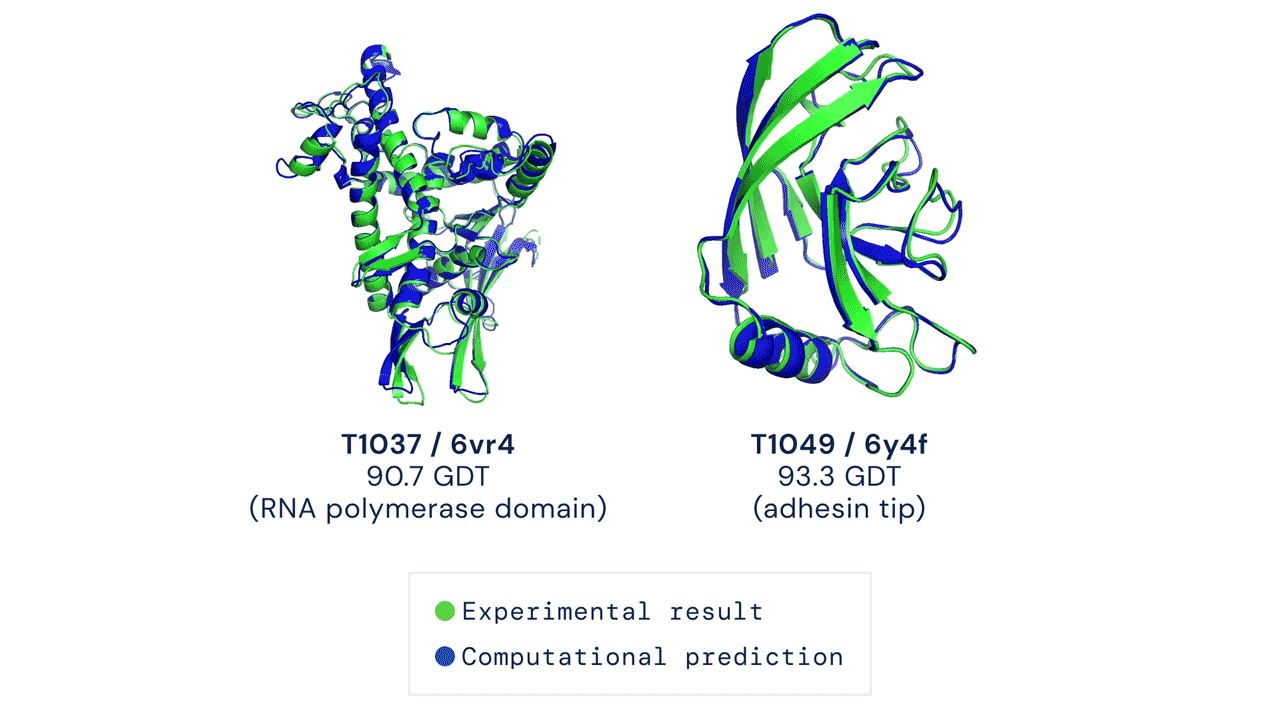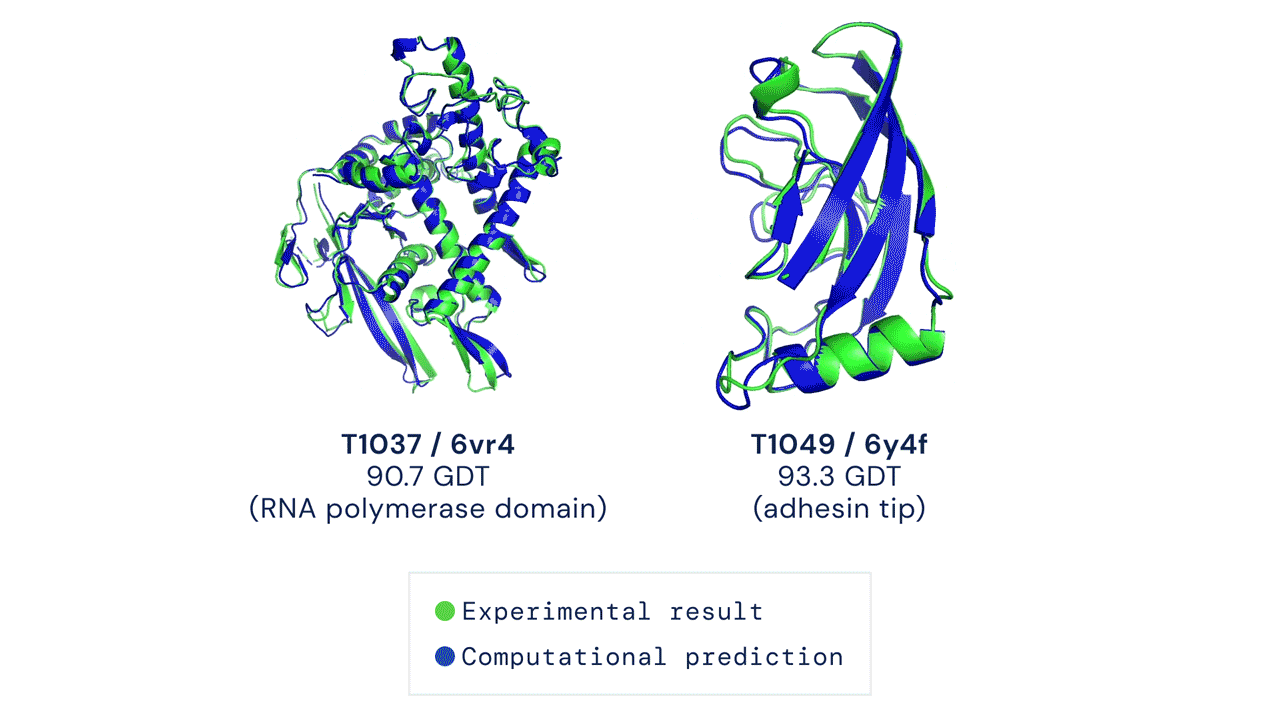
Áttörést hoz a biológiába a minden eddiginél pontosabb fehérjekutató algoritmus, az AlphaFold2
A Google-féle DeepMind legújabb AI-modellje az aminosavak sorrendjéből egész pontosan megfejti a fehérjék háromdimenziós térszerkezetét. A mesterséges intelligencia forradalmasíthatja a gyógyszerkutatást: van olyan rákkutató cég, ahol az AlphaFold2 a korábbi egy hónapról néhány órára csökkentette a hatóanyag-jelölt fehérjék megtalálását.

Öntudatra ébredt a mesterséges intelligencia? Veszélyben érzi magát? Ügyvédet fogadott? Mi folyik a Google-nél?
A Google június közepén kényszerszabadságra küldte Blake Lemoine-t, a szoftvermérnököt, aki érző lénynek nevezte a cég legújabb AI-fejlesztéseiből született LaMDA-t. A chatbot ezután állítólag ügyvédet kért, és Lemoine szerzett is neki egyet. Mindeközben a mesterséges intelligenciát kutató szakértők mellett filozófusok is vitatják, hogy a számítógépes modell emberi tulajdonságokkal bírna.
Több mint 12 ezer eddig ismeretlen mikroorganizmust azonosítottak
Amerikai kutatók több mint 52 ezer élőlény genomját határozták meg, és 12 ezer új mikrobafajt fedeztek fel, ami 44 százalékkal terjeszti ki az eddig ismert baktériumok és archeák közé tartozó mikroorganizmusok mennyiségét.
Kapcsolódó cikkek