Ez a szerkezet 700 kilót nyom, akkora, mint egy hűtőszekrény, és olyasmire képes, amiről 20 éve még csak álmodtunk

Közel két évtizeddel azután, hogy kutatók az 1950-es években megfejtették a DNS szerkezetét, megjelentek az első olyan módszerek, amelyek képesek voltak feltárni a földi élet fő információtároló molekulája által hordozott genetikai utasításokat. A kettős hélixet alkotó két nukleotidláncban az információt a négy nukleobázis – az adenin, timin, citozin és guanin – sorrendje rejti, aminek a kiolvasását DNS-szekvenálásnak nevezzük.
A szekvenálási technológiák az első emberi genom 2003-ban történő összeállítása óta robbanásszerű fejlődésen mentek keresztül, ami korábban elképzelhetetlen mennyiségű genetikai adatot tett elérhetővé a kutatóknak és a klinikai szakembereknek. Amellett, hogy emiatt saját magunkat és bolygónk többi élőlényét is sokkal jobban megismertük, a szekvenálási kapacitás bővülése és költségének meredek zuhanása az egészségügyet is elkezdte átformálni. Egyre elérhetőbbé válik a személyre szabott orvoslás, és ma már néhány nap alatt hatásos vakcinákat tudunk kifejleszteni új kórokozók ellen.

Az nem kétséges, hogy a DNS-szekvenálás egyre nagyobb szerepet játszik majd a kutatásban, az egészségügyben és az iparban, az alkalmazások száma pedig csak nő a tudományos közösség kreativitásával és innovációjával. De hogy jutottunk el néhány évtized alatt az apró vírusgenomok nehézkes és költséges meghatározásától oda, hogy ma egyetlen szekvenáló készülék 1-2 nap alatt akár 128 ember teljes genomját is feltárja? És mit hozhat a jövő?
Egy vírus DNS-szekvenciájától az első emberi genomig
Az első DNS-szekvenálási technológia kifejlesztése a Kínában született amerikai genetikus, Ray Wu nevéhez fűzödik, aki módszerével 1970-ben, a Cornell Egyetemen meghatározta egy baktériumokat fertőző vírus (bakteriofág) DNS örökítőanyagának egy apró részletét. Később, 1975-ben Frederick Sanger brit biokémikus közölte az első, valóban hatékony DNS-szekvenálási módszert, amellyel majdnem elkészítették egy bakteriofág több mint 5 ezer nukleotidból álló genomját. Sanger, akinek két kémiai Nobel-díjat is megítéltek, kollégáival együtt egy új módszert fejlesztett ki két évvel később, amit a kutatóról, Sanger szekvenálásnak neveztek el. Sangerrel nagyjából egy időben publikálták saját módszerüket Allan Maxam és Walter Gilbert amerikai molekuláris biológusok – ez Maxam–Gilbert szekvenálásként vált ismertté.

A Sanger-féle szekvenálás Richard McCombie és kollégáinak 2019-es, szekvenálási technológiákat áttekintő tanulmánya szerint lényegében leutánozza azt, ahogy a sejtjeinkben a DNS másolása (vagy replikációja) lezajlik. A módszer a DNS-szintézist megállító, kezdetben radioaktív izotópokkal ellátott dideoxi-nukleotidok beépítésén és azok filmeken történő észlelésén alapult, amit idővel fluoreszcens jelölés és lézeres észlelés váltott fel. A kutatók szerint ahhoz, hogy tényleg beköszöntsön a genomika korszaka, kellett még egy dolog: az a szoftvercsomag, amit az angliai Medical Research Council (MRC) molekuláris biológiai laboratóriumában Rodger Staden fejlesztett ki az 1980-as években – ez tette lehetővé az első genomok elkészítéséhez elengedhetetlen szekvenciaillesztést is.

Az első teljes egészében megszekvenált genom az MS2 bakteriofág több mint 3 ezer nukleotidból álló RNS-szekvenciája volt, amit Walter Fiers belga molekuláris biológusnak és kollégáinak sikerült elkészíteni 1976-ban. Az első élőlény, az emberi fertőzéseket is okozó Haemophilus influenzae baktérium genomjára majdnem 20 évet kellett még várni, de ezután felpörögtek az események: 1996-ban elkészült az első eukarióta, a sörélesztő (Saccharomyces cerevisiae) genomja, majd az első állat, aminek a genomját meghatározták, a genetikai modellélőlényként ismert Caenorhabditis elegans fonálféreg lett, 1998-ban.
Eközben 1990-ben az amerikai Energiaügyi Minisztérium (Department of Energy, DOE) és a Nemzeti Egészségügyi Intézet (National Institute of Health, NIH) elindította az emberi genom projektet (Human Genome Project), amely 11 évvel később nemzetközi együttműködésben előállt az első kezdeti (draft) emberi genommal. A 2001-ben publikált szekvenciát egy teljesebb, a genom nagyjából 90 százalékát lefedő követte, és ezzel a projektet az NIH befejezettnek nyilvánította – bár valójában a komplett emberi genom szekvenciájára egészen 2023 augusztusáig kellett várni.
Az NGS-forradalom, ami exponenciálisan csökkenti a költségeket
Az első emberi genomszekvencia elkészítését Shawn Levy és Braden Boone, az amerikai HudsonAlpha biotechnológiai intézet kutatóinak átfogó tanulmánya szerint három alapvető technológiai újítás tette lehetővé: a DNS-minták sokszorosítására használt polimeráz láncreakció (polymerase chain reaction, PCR) kifejlesztése az 1980-as évek végén; a magas minőségű nukleinsav-módosító enzimek elérhető válása széles körben; valamint az automatizált, Sanger-alapú fluoreszcens DNS-szekvenálás kifejlesztése.
A 2000-es évek közepétől új szekvenálási technológiák jelentek meg, amelyek az eredetileg a Cambridge-i Egyetem kutatói, Shankar Balasubramanian és David Klenerman által kifejlesztett szintézisen alapuló szekvenálást (sequencing by synthesis, SBS) alkalmazták. Ezeknek az új generációs szekvenálásnak (next generation sequencing, NGS) nevezett módszereknek a legáttörőbb újítása Levyék szerint az, hogy egyetlen futtatás során egyszerre több milliótól több billióig terjedő megfigyelést tesznek lehetővé. Azt állítják, elsősorban ennek a lenyűgöző mértékű párhuzamosításnak köszönhető az, hogy az utóbbi években exponenciálisan nőtt a szekvenálással generált adatmennyiség, miközben a költségek is exponenciálisan csökkentek.
„Az NGS technológia által lehetővé tett szekvenálási áteresztőképesség [az egyszerre leolvasott DNS-molekulák száma] ugrásszerű növekedése drámaian megváltoztatta a genomunkról és magunkról alkotott képünket” – írják McCombie-ék a tanulmányukban. A szekvenálás során egy adott DNS-darabról (fragment) leolvasott szekvenciát a genetikusok readeknek nevezik, amiket hosszúságuk alapján rövid (short-read) vagy hosszú (long-read) readekre különítenek el. Az NGS szekvenálás maximum pár száz bázispár hosszú, vagyis short-read szekvenálási technológia – ennek előnye, hogy az általa generált adatok könnyebben értelmezhetők, legalábbis ha már rendelkezésre áll egy referenciagenom, amihez hozzá lehet őket illeszteni (alignment). Ezután algoritmusokkal már viszonylag könnyű egyetlen nukleotid módosulásával járó egypontos nukleotid-polimorfizmusokat (single nucleotide polymorphism, SNP) azonosítani a vizsgált és a referencia emberi genom között.
Az első forgalomba hozott NGS-berendezés az amerikai 454 Life Sciences biotechnológiai vállalat GS20 szekvenálója volt, amit 2004-ben jelentettek be. Ezt néhány évvel később követték a szintén amerikai Illumina és Ion Torrent NGS-szekvenálói, majd a 2010-es években az Illumina vált piacvezetővé a területen. Ez annak fényében nem meglepő, hogy a 2015-ben bejelentett HiSeq X szekvenáló platformjuk Levy és Boone szerint elképesztően megnövelte az egyszerre megszekvenálható DNS mennyiségét, amire szinte rögtön több nagyszabású vizsgálatot húztak fel, köztük az 1000 Genom Projektet.

A kutatók azt állítják, az Illumina HiSeq X rendszere volt évekig az egyetlen szekvenálási technológia, ami egy emberi genom méretskáláján nagyon precíz adatokat tudott generálni, miközben a genom elkészítéséhez szükséges reagensek ára 1000 dollár alatt alakult. Aztán 2017-ben az Illumina NovaSeq 6000 platformjának piacra dobása tette lehetővé, hogy a szekvenálási kapacitás tényleg robbanásszerű növekedésnek induljon. Ezt tanulmányukban azzal illusztrálják, hogy egy 10 NovaSeq 6000-est üzemeltető szekvenáló központ évente 60 ezer 30-szoros lefedettségű (coverage) genomot tud megszekvenálni – a lefedettség a szekvencia pontosságát érzékelteti és azt fejezi ki, hogy hány egyedi read tartalmazza a szekvencia egy adott nukleotidját. Az Illumina által alkalmazott, az előkészítéstől az adatfeldolgozásig több lépésből álló szekvenálási módszert a cég videója mutatja be:
„Ahogy a genomszintű szekvenálás rutinná vált, paradigmaváltás indult be a genomikában, ami a nagy áteresztőképességű, gyors szekvenálási képességet nagymintás vizsgálatokban használja ki” – írta Levy és Boone. A szakemberek szerint ezen új megközelítéseknek közvetlen hatásai lesznek a személyre szabott orvoslásra és a földi élővilág változatosságának, evolúciójának megértésére. Az NGS technológia robbanásszerű fejlődése persze kihívásokkal is jár: nem könnyű a kutatóknak és klinikai szakembereknek lekövetni az akár havi szinten változó berendezéseket, felhasznált kémiai reagenseket és eljárásokat.
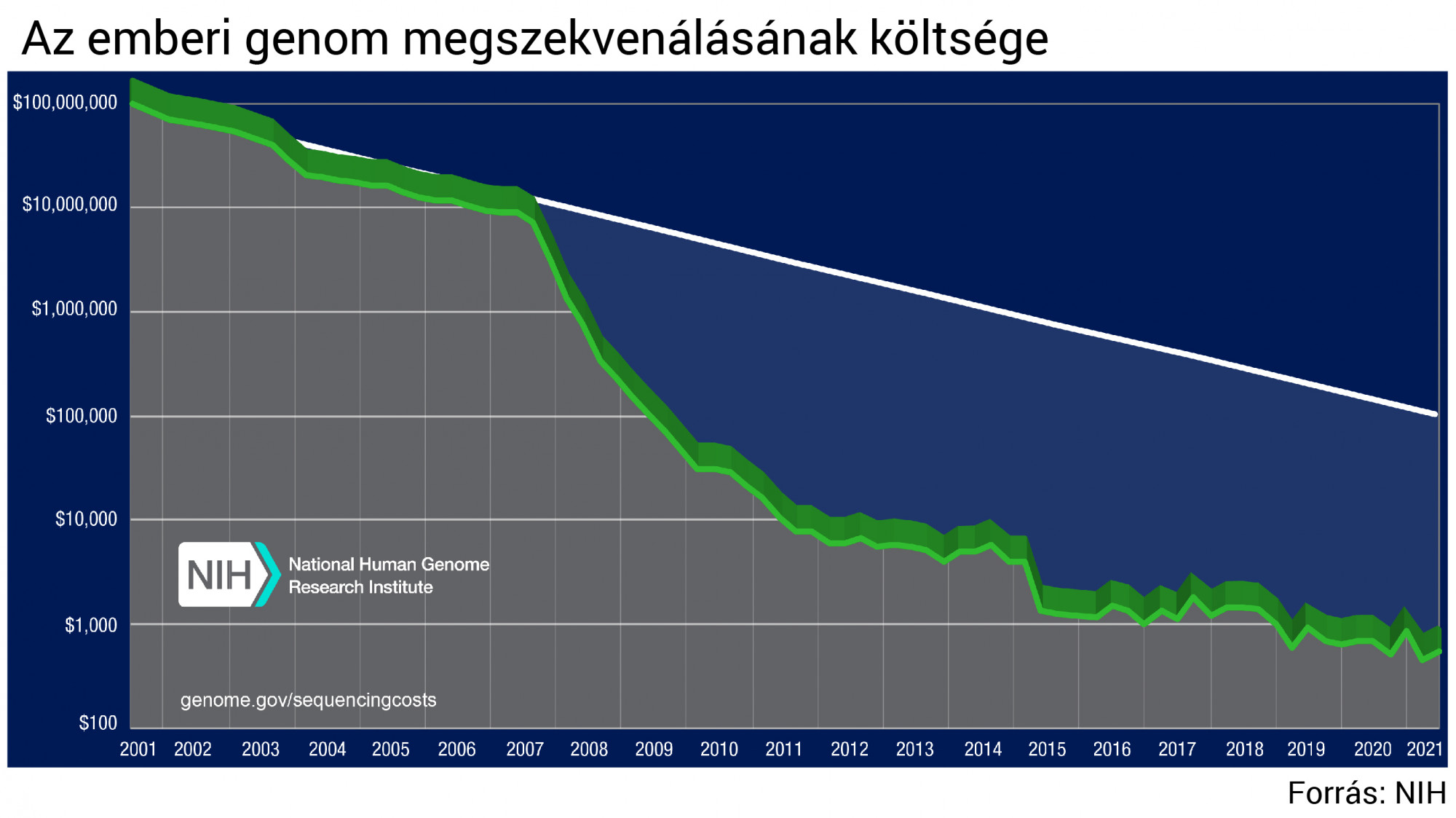
Az Illumina tavaly szeptemberben bemutatta a NovaSeq X sorozatú szekvenálóját, amit a cég az eddigi legjobb és legnagyobb áteresztőképességű szekvenáló platformjának nevez. A hűtő méretű, közel 700 kilogrammos, csúcstechnológiás short-read NovaSeq X szekvenáló egyetlen áramlási cellájával (flow cell) egy futtatás során akár 64 emberi genomot is képes meghatározni. A két áramlási cellával felszerelt NovaSeq X Plus ezt 128 emberi genomra bővíti – ezek elkészítéséhez régebben több tucat szekvenáló berendezésre lett volna szükség.

A tenyerünkben elfér a szekvenáló, de nem ez a legnagyobb előnye
Levy és Boone szerint az alacsony költségű, nagy teljesítményű szekvenálás elérhetősége tovább bővíti a genomika alkalmazási körét, míg a szekvenáló platformok fejlesztése és átdolgozása, különösen a long-read technológiák terén, kitágítja azoknak a genomi szerkezeteknek a körét, amiket fel lehet tárni. A szakemberek ezzel arra utalnak, hogy a short-read szekvenálásnak nemcsak a genomok semmiből történő összeszerelése (de novo assembly) okoz kihívást, hanem a genom komplexebb, ismétlődő szakaszokkal teli régióinak felderítése is. Erre hozhatnak megoldást a harmadik generációs, egymolekulás szekvenáláson (single molecule sequencing, SMS) alapuló technológiák, amelyek hosszú DNS-readekkel dolgoznak.
Amint Konstantina Athanasopoulou, az Athéni Nemzeti Egyetem biokémikusa és kollégái 2021-es áttekintő tanulmányukban összefoglalták, az első valóban használható long-read szekvenáló a Pacific Biosciences (PacBio) amerikai biotechnológiai vállalat 2011-ben piacra dobott RS berendezése volt. Bár ez még viszonylag rövid long-readekkel és magas hibaaránnyal dolgozott, a Sequel és Sequel IIe berendezések már tízszer akkora, 15 ezer bázis hosszúságú readeket voltak képesek leolvasni, mindössze 10-15 százalékos hibaaránnyal. A PacBio szekvenálási módszerét az alábbi videó mutatja be:
Eközben egy brit biotechnológiai vállalat, az Oxford Nanopore Technologies (ONT) 2014-ben piacra dobta saját harmadik generációs szekvenáló technológiáját, ami reagensek nélküli, nanopórus alapú szekvenálást alkalmaz. A leginkább a tenyérben elférő MinION szekvenálóról ismert – és a koronavírus-járvány alatt széles körben alkalmazott – technológia lényegét egy speciális membrán adja, amelynek a nanopórusain áthaladnak a DNS- vagy RNS-molekulák. Az áthaladás során a különböző bázisok más-más elektromos jelet generálnak, ami alapján a szekvenáló szoftvere be tudja azonosítani őket, ahogy ez az ONT videójából is kiderül:
Levy és Boone szerint a MinION bejelentése számos korlátot áttört azzal, hogy ez volt az első tenyérben elférő, és működéséhez csak egy USB tápellátást igénylő szekvenáló, amely mindössze 1000 dollárba került. A cég azóta piacra dobott egyszerre több áramlási cellával dolgozó szekvenálókat, az 1-5 cellás GridION-t és az akár 48 cellás PromethION-t, amikkel már viszonylag nagy áteresztőképességet lehet elérni. Eközben az Illumina is kifejlesztette a meglévő NGS berendezésein bevethető saját long-read szekvenálási technológiáját, és más vállalatok is dolgoznak azon, hogy betörjenek a harmadik generációs szekvenálási technológia piacára.

Új távlatokat nyit, de sokat kell még fejlődnie
De miért az NGS a legelterjedtebb szekvenálási módszer továbbra is, ha már itt az újabb technológia? McCombie és kollégái szerint ennek az az elsődleges oka, hogy a long-read szekvenálás az NGS-sel összehasonlításban alacsony áteresztőképességű és magas költségű. Emellett az NGS szekvenálás további előnye, hogy kevesebb DNS-mintát igényel, így alkalmasabb az általában kevés örökítőanyagot tartalmazó klinikai minták szekvenálására. A long-read szekvenálás ma még elsősorban genomok de novo összeszerelésekor, valamint komplex, ismétlődő DNS-szakaszokkal teli genomi régiók – ilyen például az Y kromoszóma egy része – meghatározásakor jön jól.
A korlátai ellenére a long-read szekvenálást hatalmas dolognak nevezte Fergal Martin, az Európai Bioinformatikai Intézet egyik kutatócsoportját vezető szakember, aki nemrég közel egy tucat szakértővel együtt nyilatkozott a technológia jelenéről és jövőjéről a Nature Methodsnak. A folyóirat 2022-ben a long-read szekvenálást választotta az év módszerének, ami nélkül szerintük több nagy, most futó genomikai projekt elképzelhetetlen lenne. Martin elmondta, az új módszer segítségével jobban fel tudják deríteni a genomok strukturális variációit és long-read RNS-szekvenálással az éppen kifejeződő géneket is azonosítani tudják.
A long-read szekvenálás sokat fejlődött az elmúlt években, de a szakemberek szerint továbbra is több megoldandó problémával néz szembe. Hardip Patel, az Ausztráliai Nemzeti Egyetem Őslakos Genomikai központjának kutatója szerint az sem tiszta még, hogy melyik long-read szekvenálási technológia a legjobb. Patel és kollégái egy 500 őslakos ausztrál genomjából álló pángenomon dolgoznak, ami a most rendelkezésre álló genetikai adatoknál jobban reprezentálja majd az ország őslakos közösségeit. Ehhez az Y kromoszóma hiánytalan, augusztusban közölt szekvenciájának létrehozása során alkalmazott technológiákat, long-read PacBio és ONT szekvenálást használnak majd short-read Illumina szekvenálással együtt.
A kutatók most arra várnak, hogy a long-read szekvenálás pontosabbá váljon, mivel a szükséges precizitás eléréshez ma – ahogy az előbb említett két projektnél is – több platformot kell használni. Patel tapasztalatai szerint a nanopórus alapú szekvenálás precizitása folyamatosan javul: amikor tavaly elkezdtek adatokat gyűjteni a projektjükhöz, az ONT módszere még 92 százalékos pontosságnál tartott, mostanában viszont már a 99 százalékhoz közelít. Ha a következő években sikerülne elérni a kellő pontosságot és növelni a long-read technológia áteresztőképességét, a szekvenálás egyetlen platformon is elvégezhető lenne, ami tovább csökkentené annak költségeit, miközben a legkomplexebb genomi régiókkal is meg tudna birkózni.
Kapcsolódó cikkek a Qubiten:
Három dimenzióban kell gondolkodnunk, ha meg akarjuk érteni a DNS működését
A genomot sejtmagba csomagoló térbeli szerkezetet és annak működését csak mostanában kezdjük igazán megérteni, de az már látszik, hogy ennek feltárása segíthet megérteni a vírusos fertőzések lefolyását és egyes betegségek okait.

33 év után végre a kezünkben van az emberi genom teljes szekvenciája
Már csak az Y kromoszómára kellett várni, aminek szekvenciájából eddig 30 millió bázispár hiányzott. De miért tartott eddig a munka, és mi ennek a jelentősége?
A szintetikus biológia korában élünk, de a mesterséges élet létrehozása még mindig csak álom
Az elmúlt húsz év áttöréseinek köszönhetően mára lehetővé vált az élőlények, köztük az ember genomjának szerkesztése, de minden igyekezet ellenére még nagyon messze vagyunk attól, hogy teljesen szintetikus sejtet állítsunk elő.
Kapcsolódó cikkek



