Elképesztően fejlett fehérjekutató algoritmusok versengenek egymással, hogy feltérképezzék a legalapvetőbb biológiai folyamatokat

Ritka az olyan mértékű tudományos áttörés, mint amilyet 2020-ban a fehérjék háromdimenziós szerkezetét megfejtő AlphaFold2 mesterséges intelligencia fejlesztői produkáltak egy addig kevesek által ismert bioinformatikai versenyen. Az algoritmus praktikusan megoldotta, hogyan alakul ki az élethez nélkülözhetetlen, aminosav-maradékok láncolatából felépülő fehérjék háromdimenziós struktúrája, és ez egy sor biotechnológiai és gyógyszerkutatási felfedezést alapozhat meg a következő években.
A fehérjefeltekeredés problémája évtizedek óta foglalkoztatja a molekuláris biológusokat, és az 1990-es évek közepe óta léteznek olyan irányú próbálkozások, hogy egyre jobb, a fehérjeszekvenciából térbeli modelleket generáló algoritmusokat fejlesszenek ki. Ez a célja a kétévente megrendezett CASP (Critical Assessment of protein Structure Prediction) versenynek, amelynek legutóbbi körét (CASP15) tavaly decemberben zárták le egy, a törökországi Antalyában megrendezett konferencián.
De ha az AlphaFold2 gyakorlatilag megoldotta a kérdést, mi értelme újabb versenyt rendezni? Ezzel az általános vélekedéssel találkoztak a Nature szerint a CASP15 szervezői, köztük a Marylandi Egyetemen számítógépes biológiával foglalkozó John Moult, aki azt állítja, a helyzet ennek éppen a fordítottja. A CASP15-ön a legsikeresebb kutatócsoportok mind az AlphaFold2 algoritmusára építettek, annak különféle módosításával, amivel kissé jobb eredményeket produkáltak egyes fehérjék és fehérjealegységek esetén.
Annyira pontos az AlphaFold2, hogy nehéz hasonló mértékű előrelépést tenni
Az AlphaFold-forradalom kezdete 2018 végére, a CASP13 versenyre nyúlik vissza. Itt már a Google anyacégéhez, az Alphabethez tartozó DeepMind vállalat mélytanulási (deep learning) algoritmusa bizonyult a legjobbnak, de úgy tűnt, a kutatásokhoz és biotechnológiai alkalmazásokhoz szükséges pontosságra még sokáig várni kell. Nem így alakult: két év múlva, a CASP14-en megérkezett az áttörés, és az algoritmus továbbfejlesztett változata, az AlphaFold2 precizitásával lemosta a mezőnyt.
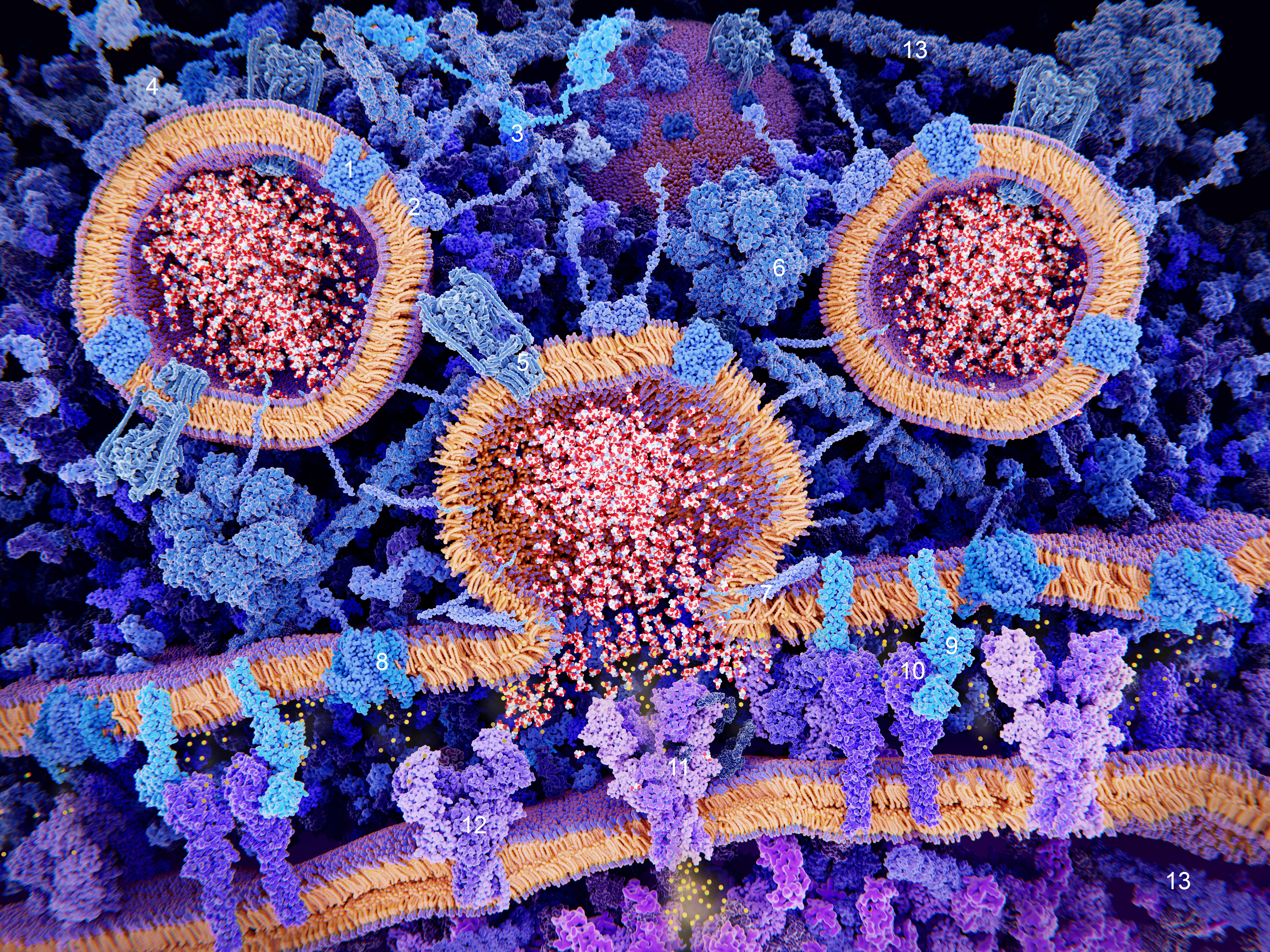
2021 nyarán aztán a DeepMind kutatói a Nature-ben publikált tanulmányukban részletesen ismertették az AlphaFold2-t, valamint az emberi sejtek által kifejezett közel 21 ezer fehérje (emberi proteom) háromdimenziós szerkezetét, és a GitHub-on mindenkinek elérhetővé tették a mélytanulási algoritmust. Az AlphaFold2 azóta tízezerszer ennyi, 200 millió fehérje térszerkezetét határozta meg – ezek mind nyíltan elérhetők az európai bioinformatikai intézet (EBI) és a DeepMind által kifejlesztett adatbázisban.
A CASP15-ön, amelyen a DeepMind ezúttal nem vett részt, az eredménytáblázat szerint a 162 kutatócsoport közül a kínai Nankai Egyetem szakembere, Jang Csien-ji által vezetett Jang-Server kutatócsoport teljesített a legjobban, legalábbis a meghatározott térszerkezetek precizitását kifejező Z-pontszámok összesítése alapján. Ez a CASP kísérletekhez használt, strukturális adatbázisokban nem szereplő fehérjékhez és RNS molekulákhoz legenerált 3D modelleket veti össze azok kísérleti úton, kriogén-elektronmikroszkópiával vagy röntgen-krisztallográfiával meghatározott szerkezetével.
Jang és munkatársai az AlphaFold2 neurális hálójának egyik fő, Evofomernek nevezett komponensét integrálták trRosettaX2 fehérjekutató algoritmusukkal. Így 90,4-es Z-pontszámot értek el, amivel kissé megelőzték az UM-TBM és PEZYFoldings csoportokat. Jangék sikerének titka az lehetett Sergey Ovchinnikov, a Hardvard Egyetem molekuláris biológusa szerint, hogy a fehérjestruktúrákról evolúciós mintázatok alapján információt hordozó, úgynevezett többszörös szekvenciaillesztésekhez (multiple sequence alignment) még több adatot nyertek ki genetikai adatbázisokból, különösen vírusfehérjék esetén, amelyeknél kevés evolúciósan rokon szekvencia állt rendelkezésre.

Ovchinnikov úgy látja, hogy mivel a Baker Lab csoport RosettaFold2 alapú szoftverén kívül a legjobban teljesítő algoritmusok valamilyen szinten kivétel nélkül az AlphaFold2-t használták, visszatértünk oda, hogy minden azon múlik, ki tudja a legjobb többszörös szekvenciaillesztést létrehozni. A CASP15-ön versenyző algoritmusok – az eredmények kiértékelésében résztvevő Daniel Rigden bioinformatikus szerint – lenyűgöző sikereket értek el a molekulaszerkezetek meghatározásában, néhány érdekesebb kudarccal együtt. Ilyen volt két, levéltetvek sejtjei által kifejezett, viszonylag apró és másodlagos szerkezetük alapján alfa-helikális fehérje, a T1130 és T1131, amelyekből csak előbbinél tudott a Jang-Server algoritmus viszonylag jó megoldást adni.
Mi a következő lépés?
A CASP15 szervezői részben korábbi tesztek megváltoztatásával, részben újak létrehozásával reagáltak az AlphaFold2 utáni világra. Ezekben már a fehérjék más molekulákkal, például gyógyszerek hatóanyagaival történő interakcióit vizsgálták, vagy az egyes fehérjék által felvehető különböző térbeli szerkezeteket fejtették meg.
Az AlphaFold2 módosításával a kutatók azt is el tudták érni, hogy az algoritmus a több, egymással kölcsönható fehérje által alkotott fehérjekomplexek szerkezetét is meghatározza. Ezek a komplexek vírusos fertőzéseknél lejátszódó immunológiai folyamatok mellett olyan alapvető sejtbeli mechanizmusokban játszanak szerepet, mint a jelátviteli útvonalak, a DNS RNS-re átírása (transzkripció), vagy a hírvivő RNS (mRNS) fehérjékké való lefordítása (transzláció).
A CASP15 során az algoritmusok a korábbiaknál sokkal pontosabban adták vissza a komplexek szerkezetét, és ebben a nagyobb adathalmazok mellett olyan neurálisháló-komponensek segítették őket, mint az AlphaFold2 Evoformerje. A Nature-nek nyilatkozó Arne Elofsson, a Stockholmi Egyetem bioinformatikusa szerint e komplexek precíz szerkezetének feltárása fontos új kihívást jelent, mert ezen a téren bőven van még az algoritmusoknak hová fejlődni.
Kapcsolódó cikkek a Qubiten:
KAPCSOLÓDÓ CIKKEK
Áttörést hoz a biológiába a minden eddiginél pontosabb fehérjekutató algoritmus, az AlphaFold2
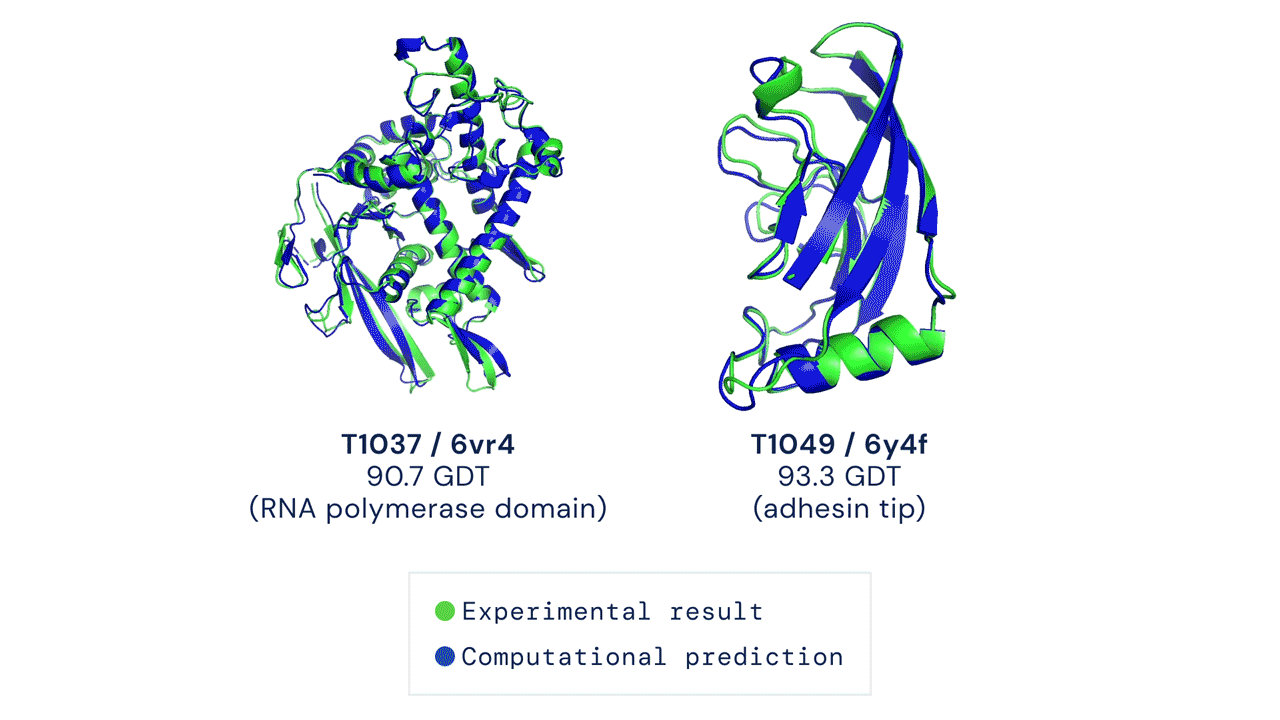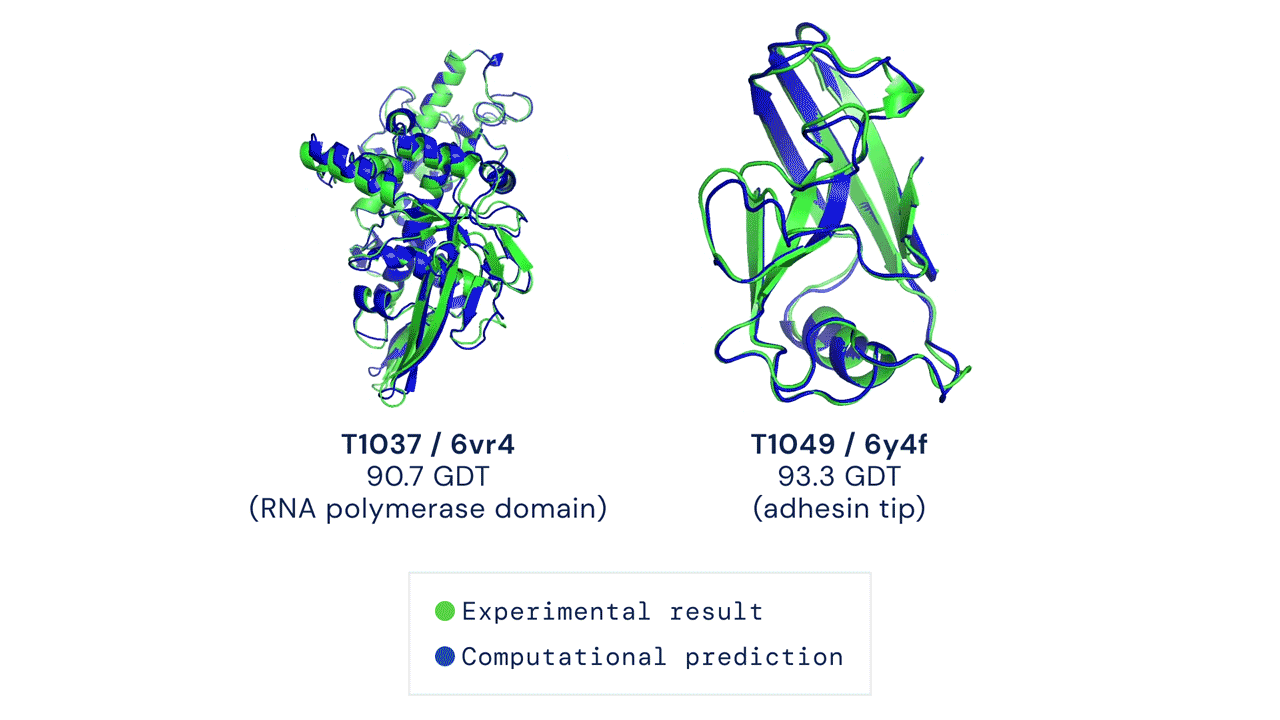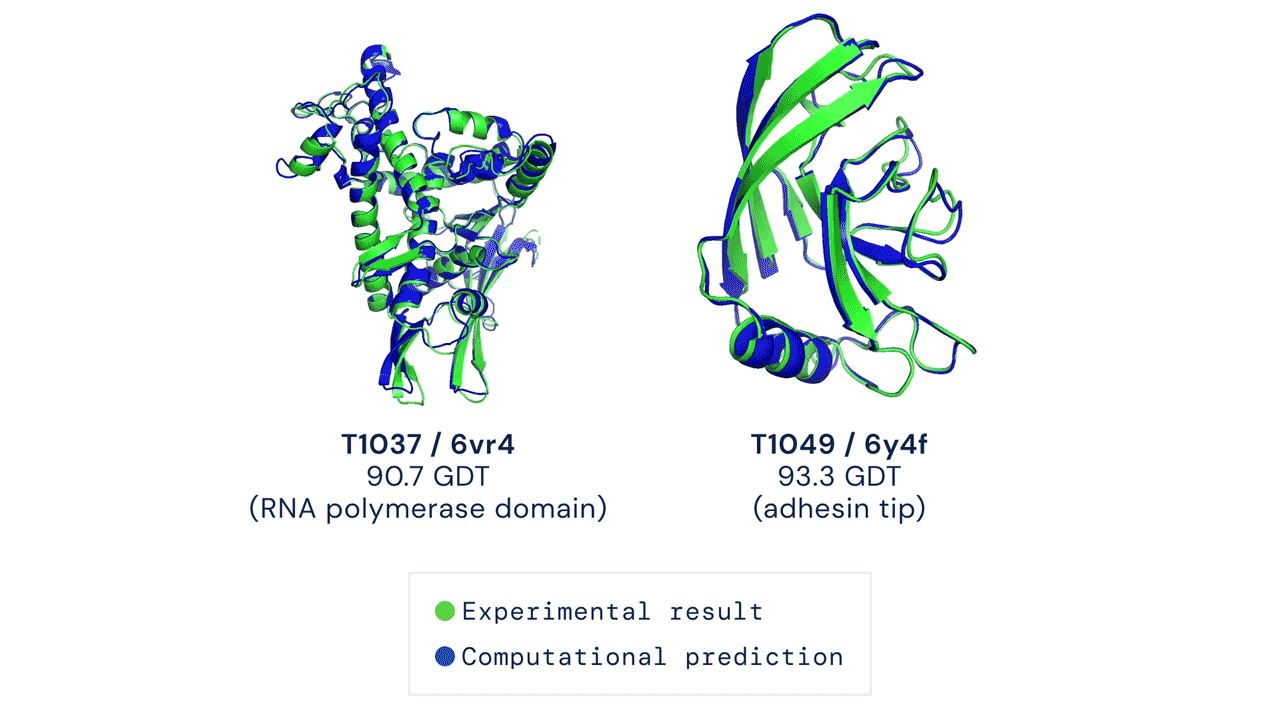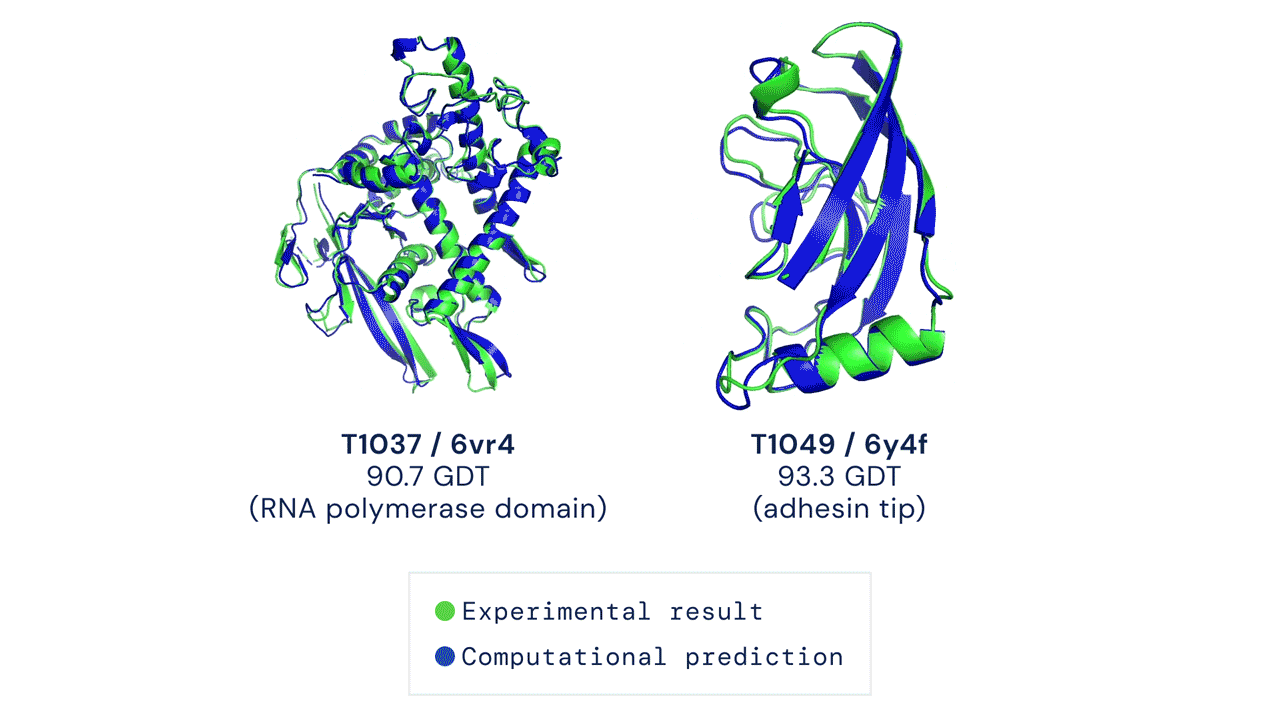

Fél évszázados problémát oldott meg, egy csapásra forradalmasíthatja a biológiát a DeepMind algoritmusa
200 millió fehérje térszerkezetét tárta fel egyetlen év alatt a DeepMind fehérjekutató algoritmusa